
Neuromuscular Connectivity in health and diseases
Our laboratory aims to understand the cellular and molecular mechanisms underlying neuromuscular synapse assembly and maintenance in physiological and pathological conditions.
The Neuromuscular junction (NMJ) is the contact zone between motor neurons and skeletal muscles. This synapse drives the precise initiation and control of motion. Therefore, much of our behavior and wellness relies on the appropriate functioning of this specialised structure. Neuromuscular transmission deficiency occurs in a large array of rare human diseases including channelopathies, congenital or acquired myasthenia and amyotrophic lateral sclerosis. Most of these pathologies are untreatable and life threatening with devastating economic and societal consequences in terms of loss of quality of life and of the burden of disability. The patients display complex clinical phenotypes mainly characterized by a profound muscle weakness and loss of mobility.
We combine unbiased screens and a large array of functional assays including quantitative morphological imaging, behavioral analysis and electrophysiology using mouse models and/or human-derived specimens to explore the complexity of the trans-synaptic mechanisms controlling neuromuscular connectivity. Our overarching goal is to improve physiopathological knowledge that can be used not only for molecular diagnosis and genetic counselling of families affected with the diseases of interest, but also to design new targets of therapeutic interest.
To achieve this goal, our team is built on an organization that favors tight interactions between practicing neurologists from the Paris Est French reference center for neuromuscular diseases and researchers/research assistants together with a large network of national/international collaborations to share knowledge and expertise.
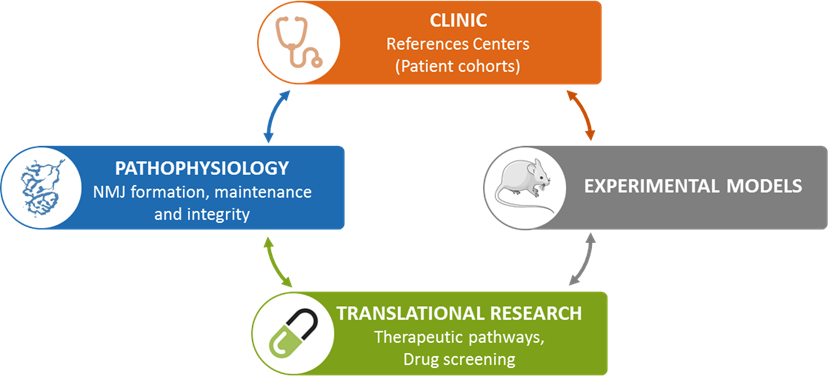
We defined three main aims of research:
1) Characterize the molecular determinants underlying NMJ assembly and maintenance (PI: Laure Strochlic/Julien Messéant).
We have recently identified a new trans-synaptic pathway at the NMJ and developed innovative tools using mouse genetics to dissect its molecular characteristics.
2) Understand how disruption of nerve/muscle communication leads to neuromuscular diseases such as myasthenia and amyotrophic lateral sclerosis (PI: Stéphanie Godard-Bauché/Gaelle Bruneteau).
Thanks to our clinical expertise and our national networks, we analyze the physiopathological mechanisms underlying the studied diseases in patients.
3) Modulate trans-synaptic function to restore appropriate synaptic connectivity in a pathological context as a basis for therapeutic interventions (PI: Bertrand Fontaine/Laure Strochlic).
We develop innovative pharmacological or genetic strategies that promote trans-synaptic communication and nerve/muscle attachment to mitigate NMJ disease symptoms to ultimately prevent or compensate the progression of the loss of motor function in neuromuscular diseases.
Key words: Neuromuscular Junction, Neuromuscular disorders, disease modeling mouse models, human derived specimens, quantitative imaging, therapeutic strategies.
Main collaborations:
- Rozen Le Panse and Sonia Berrih-Aknin (France)
- Arnaud Ferry (France)
- Cécile Martinat (France)
- Eric Krejci (France)
- Frédérique Charbonnier (France)
- Nathalie Sans and Mireille Montcouquiol (France)
- Laurent Schaeffer, NeuroMyoGene Institute, (France)
- Hanns Lochmüller, The Children’s Hospital of Eastern Ontario (Canada)
- Markus Ruëgg, Biozentrum (Switzerland)
- Anna Punga, Uppsala University, (Sweden)



| Name | Position | ORCID |
|---|


Journal articles
- Savine Vicart, Yann Péréon, K. Ghorab, A. Pegat, R. Dufresne, et al.. Self-reported outcomes and quality of life of patients with non-dystrophic myotonia: The French IMPACT 2022 survey.. Revue Neurologique, 2024, Revue Neurologique, Online ahead of print. ⟨10.1016/j.neurol.2024.04.007⟩. ⟨hal-04613788⟩
- Marion Masingue, Olivia Cattaneo, Nicolas Wolff, Céline Buon, Damien Sternberg, et al.. New mutation in the β1 propeller domain of LRP4 responsible for congenital myasthenic syndrome associated with Cenani–Lenz syndrome. Scientific Reports, 2023, 13 (1), pp.14054. ⟨10.1038/s41598-023-41008-5⟩. ⟨hal-04191765⟩
- Tanya Stojkovic, Marion Masingue, Helène Turmel, Marianne Hezode-Arzel, Anthony Béhin, et al.. Diagnostic yield of a practical electrodiagnostic protocol discriminating between different congenital myasthenic syndromes. Neuromuscular Disorders, 2022, 32 (11-12), pp.870-878. ⟨10.1016/j.nmd.2022.10.001⟩. ⟨hal-04074000⟩
- Arnaud Jacquier, Valérie Risson, Thomas Simonet, Florine Roussange, Nicolas Lacoste, et al.. Severe congenital myasthenic syndromes caused by agrin mutations affecting secretion by motoneurons. Acta Neuropathologica, 2022, 144 (4), pp.707-731. ⟨10.1007/s00401-022-02475-8⟩. ⟨hal-03863096⟩
- laure strochlic. Entre muscle et nerf: comprendre la jonction neuromusculaire pour mieux comprendre le mouvement. The Conversation France, 2022. ⟨inserm-03774979⟩
- Myriam Boëx, Steve Cottin, Marius Halliez, Stéphanie Bauché, Céline Buon, et al.. The cell polarity protein Vangl2 in the muscle shapes the neuromuscular synapse by binding to and regulating the tyrosine kinase MuSK. Science Signaling, 2022, 15 (734), pp.eabg4982. ⟨10.1126/scisignal.abg4982⟩. ⟨inserm-03768653⟩
- Laura Le Gall, William J Duddy, Cecile Martinat, Virginie Mariot, Owen Connolly, et al.. Muscle cells of sporadic amyotrophic lateral sclerosis patients secrete neurotoxic vesicles. Journal of Cachexia, Sarcopenia and Muscle, 2022, 13 (2), pp.1385 - 1402. ⟨10.1002/jcsm.12945⟩. ⟨hal-04767184⟩
- Konstantinos Savvatis, Christoffer Rasmus Vissing, Lori Klouvi, Anca Rezeda Florian, Mehjabin Rahman, et al.. Cardiac Outcomes in Adults With Mitochondrial Diseases. Journal of the American College of Cardiology, 2022, 80 (15), pp.1421-1430. ⟨10.1016/j.jacc.2022.08.716⟩. ⟨hal-03837169⟩
- Tomoya Kubota, Fenfen Wu, Savine Vicart, Maki Nakaza, Damien Sternberg, et al.. Hypokalaemic periodic paralysis with a charge-retaining substitution in the voltage sensor. Brain Communications, 2021, 2 (2), ⟨10.1093/braincomms/fcaa103⟩. ⟨hal-03464510⟩
- Kevin Carvalho, Elodie Martin, Aurélia Ces, Nadège Sarrazin, Pauline Lagouge-Roussey, et al.. P2X7-deficiency improves plasticity and cognitive abilities in a mouse model of Tauopathy. Progress in Neurobiology, 2021, 206, pp.102139. ⟨10.1016/j.pneurobio.2021.102139⟩. ⟨inserm-03366671⟩
- Lucie Isoline Pisella, Sara Fernandes, Guilhem Sole, Tanya Stojkovic, Celine Tard, et al.. A multicenter cross-sectional French study of the impact of COVID-19 on neuromuscular diseases. Orphanet Journal of Rare Diseases, 2021, 16 (1), pp.450. ⟨10.1186/s13023-021-02090-y⟩. ⟨hal-03470914⟩
- Savine Vicart, Jérôme Franques, Françoise Bouhour, Armelle Magot, Yann Péréon, et al.. Efficacy and safety of mexiletine in non-dystrophic myotonias: A randomised, double-blind, placebo-controlled, cross-over study. Neuromuscular Disorders, 2021, ⟨10.1016/j.nmd.2021.06.010⟩. ⟨hal-03408750⟩
- Jean-François Desaphy, Concetta Altamura, Savine Vicart, Bertrand Fontaine. Targeted Therapies for Skeletal Muscle Ion Channelopathies: Systematic Review and Steps Towards Precision Medicine. Journal of Neuromuscular Diseases, 2021, 8 (3), pp.357-381. ⟨10.3233/JND-200582⟩. ⟨hal-03449042⟩
- Ruth Herbst, Inga Koneczny, Hanns Lochmüller, Laure Strochlic. Editorial: Molecular Mechanisms Underlying Assembly and Maintenance of the Neuromuscular Junction. Frontiers in Molecular Neuroscience, 2021, 14, pp.797832. ⟨10.3389/fnmol.2021.797832⟩. ⟨hal-03475030⟩
- Giorgia Querin, Timothée Timothée Lenglet, Rabab Debs, Tanya Stojkovic, Anthony Behin, et al.. Development of new outcome measures for adult SMA type III and IV: a multimodal longitudinal study. Journal of Neurology, 2021, 268 (5), pp.1792-1802. ⟨10.1007/s00415-020-10332-5⟩. ⟨hal-03230903⟩
- Stéphanie Bauché, Alain Sureau, Damien Sternberg, John Rendu, Céline Buon, et al.. New recessive mutations in SYT2 causing severe presynaptic congenital myasthenic syndromes. Neurology Genetics, 2020, 6 (6), pp.e534. ⟨10.1212/NXG.0000000000000534⟩. ⟨inserm-03555554⟩
- Federica Cacciamani, Luisa Sambati, Marion Houot, Marie-Odile Habert, Bruno Dubois, et al.. Awareness of cognitive decline trajectories in asymptomatic individuals at risk for AD. Alzheimer's Research and Therapy, 2020, 12 (1), pp.129. ⟨10.1186/s13195-020-00700-8⟩. ⟨hal-02982638⟩
- Antonio Atalaia, Rachel Thompson, Alberto Corvo, Leigh Carmody, Davide Piscia, et al.. A guide to writing systematic reviews of rare disease treatments to generate FAIR-compliant datasets: building a Treatabolome. Orphanet Journal of Rare Diseases, 2020, 15 (1), pp.206. ⟨10.1186/s13023-020-01493-7⟩. ⟨hal-02935405⟩
- Adèle Hesters, Maria del Mar Amador, Rabab Debs, Nadine Le Forestier, Timothée Lenglet, et al.. predictive factors for prognosis after gastrostomy placement in routine non-invasive ventilation users ALS patients. Scientific Reports, 2020, 10 (1), pp.15117. ⟨10.1038/s41598-020-70422-2⟩. ⟨hal-02969094⟩
- Bas C Stunnenberg, Samantha Lorusso, W. David David Arnold, Richard J Barohn, Stephen C Cannon, et al.. Guidelines on clinical presentation and management of nondystrophic myotonias. Muscle & nerve. Supplement., 2020, 62 (4), pp.430 - 444. ⟨10.1002/mus.26887⟩. ⟨hal-03461673⟩
- Paloma Gómez-Fernández, Aitzkoa Lopez de Lapuente Portilla, Ianire Astobiza, Jorge Mena, Andoni Urtasun, et al.. The Rare IL22RA2 Signal Peptide Coding Variant rs28385692 Decreases Secretion of IL-22BP Isoform-1, -2 and -3 and Is Associated with Risk for Multiple Sclerosis. Cells, 2020, 9 (1), pp.175. ⟨10.3390/cells9010175⟩. ⟨hal-03464472⟩
- Capucine Morelot-Panzini, Gaëlle Bruneteau, Jésus Gonzalez-Bermejo. NIV in amyotrophic lateral sclerosis: The ‘when’ and ‘how’ of the matter. Respirology, 2019, 24 (6), pp.521-530. ⟨10.1111/resp.13525⟩. ⟨hal-02365929⟩
- Raquel Guimaraes-Costa, Thomas Similowski, Isabelle Rivals, Capucine Morélot-Panzini, Marie-Cécile Nierat, et al.. Human diaphragm atrophy in amyotrophic lateral sclerosis is not predicted by routine respiratory measures. European Respiratory Journal, 2019, 53 (2), pp.1801749. ⟨10.1183/13993003.01749-2018⟩. ⟨hal-02543564⟩
- Elodie Martin, Jean Kanellopoulos, Bertrand Fontaine, Benoît Delatour, Cécile Delarasse. Le récepteur P2X7, une nouvelle cible thérapeutique dans la maladie d’Alzheimer. Médecine/Sciences, 2019, 35 (2), pp.97-99. ⟨10.1051/medsci/2019017⟩. ⟨hal-02969893⟩
- Giulia Coarelli, Rebecca Schüle, Bart P.C. van de Warrenburg, Peter de Jonghe, Claire Ewenczyk, et al.. Loss of paraplegin drives spasticity rather than ataxia in a cohort of 241 patients with SPG7. Neurology, 2019, 92 (23), pp.e2679-e2690. ⟨10.1212/WNL.0000000000007606⟩. ⟨hal-02171988⟩
- Sandra Vukusic, Romain Casey, Fabien Rollot, Bruno Brochet, Jean Pelletier, et al.. Observatoire Français de la Sclérose en Plaques (OFSEP): A unique multimodal nationwide MS registry in France. Multiple Sclerosis Journal, 2018, 26 (1), pp.118-122. ⟨10.1177/1352458518815602⟩. ⟨hal-02007918⟩
- Mitja Mitrovič, Nikolaos A Patsopoulos, Ashley H Beecham, Theresa Dankowski, An Goris, et al.. Low-Frequency and Rare-Coding Variation Contributes to Multiple Sclerosis Risk. Cell, 2018, 175 (6), pp.1679 - 1687.e7. ⟨10.1016/j.cell.2018.09.049⟩. ⟨hal-03464593⟩
- Myriam Boëx, Julien Messéant, Stéphanie Bauché, Bertrand Fontaine, Claire Legay, et al.. Regulation of mammalian neuromuscular junction formation and maintenance by Wnt signaling. Current Opinion in Physiology, 2018, 4, pp.88-95. ⟨10.1016/j.cophys.2018.07.002⟩. ⟨hal-04016110⟩
- Hugo Poulin, Pascal Gosselin-Badaroudine, Savine Vicart, Karima Habbout, Damien Sternberg, et al.. Substitutions of the S4DIV R2 residue (R1451) in NaV1.4 lead to complex forms of paramyotonia congenita and periodic paralyses. Scientific Reports, 2018, 8, pp.2041. ⟨10.1038/s41598-018-20468-0⟩. ⟨hal-01717643⟩
- Yosuke Kokunai, Carine Dalle, Savine Vicart, Damien Sternberg, Valérie Pouliot, et al.. A204E mutation in Nav1.4 DIS3 exerts gain- and loss-of-function effects that lead to periodic paralysis combining hyper- with hypo-kalaemic signs. Scientific Reports, 2018, 8, pp.16681. ⟨10.1038/s41598-018-34750-8⟩. ⟨hal-01922467⟩
- Élodie Martin, Majid Amar, Carine Dalle, Ihsen Youssef, Céline Boucher, et al.. New role of P2X7 receptor in an Alzheimer's disease mouse model. Molecular Psychiatry, In press, ⟨10.1038/s41380-018-0108-3⟩. ⟨hal-01900819⟩
- Elisa Teyssou, Laura Chartier, Maria-Del-Mar Amador, Roselina Lam, Géraldine Lautrette, et al.. Novel UBQLN2 mutations linked to Amyotrophic Lateral Sclerosis and atypical Hereditary Spastic Paraplegia phenotype through defective HSP70-mediated proteolysis. Neurobiology of Aging, 2017, 58, pp.239.e11-239.e20. ⟨10.1016/j.neurobiolaging.2017.06.018⟩. ⟨hal-03001781⟩
- Stéphanie Bauché, Geoffroy Vellieux, Damien Sternberg, Marie-Joséphine Fontenille, Elodie de Bruyckere, et al.. Mutations in GFPT1-related congenital myasthenic syndromes are associated with synaptic morphological defects and underlie a tubular aggregate myopathy with synaptopathy. Journal of Neurology, 2017, 264 (8), pp.1791-1803. ⟨10.1007/s00415-017-8569-x⟩. ⟨hal-01653176⟩
- Sophie Nicole, Yoshiteru Azuma, Stéphanie Bauché, Bruno Eymard, Hanns Lochmuller, et al.. Congenital Myasthenic Syndromes or Inherited Disorders of Neuromuscular Transmission: Recent Discoveries and Open Questions. Journal of Neuromuscular Diseases, 2017, 4 (4), pp.269-284. ⟨10.3233/JND-170257⟩. ⟨hal-01653365⟩
- Julien Messéant, Jérôme Ezan, Perrine Delers, Konstantin Glebov, Carmen Marchiol, et al.. Wnt proteins contribute to neuromuscular junction formation through distinct signaling pathways. Development (Cambridge, England), 2017, 144 (9), pp.1712 - 1724. ⟨10.1242/dev.146167⟩. ⟨hal-01542853⟩
- Stéphanie Bauché, Seana O’regan, Yoshiteru Azuma, Fanny Laffargue, Grace Mcmacken, et al.. Impaired Presynaptic High-Affinity Choline Transporter Causes a Congenital Myasthenic Syndrome with Episodic Apnea. American Journal of Human Genetics, 2016, 99 (3), pp.753 - 761. ⟨10.1016/j.ajhg.2016.06.033⟩. ⟨hal-01680226⟩
- Elodie Martin, Céline Boucher, Bertrand Fontaine, Cécile Delarasse. Distinct inflammatory phenotypes of microglia and monocyte-derived macrophages in Alzheimer's disease models: effects of aging and amyloid pathology. Aging Cell, 2016, ⟨10.1111/acel.12522⟩. ⟨hal-01379146⟩
- Karima Habbout, Hugo Poulin, Francois Rivier, Serena Giuliano, Damien Sternberg, et al.. A recessive Na v 1.4 mutation underlies congenital myasthenic syndrome with periodic paralysis. Neurology, 2016, 86 (2), pp.161-169. ⟨10.1212/WNL.0000000000002264⟩. ⟨hal-01817830⟩
- Gaelle Bruneteau, Stéphanie Bauché, Jose-Luis Gonzalez de Aguilar, Guy Brochier, Nathalie Mandjee, et al.. Endplate denervation correlates with Nogo-A muscle expression in amyotrophic lateral sclerosis patients. Annals of Clinical and Translational Neurology, 2015, 2 (4), pp.362-372. ⟨10.1002/acn3.179⟩. ⟨hal-01118997⟩
- Julien Messéant, Alexandre Dobbertin, Emmanuelle Girard, Perrine Delers, Marin Manuel, et al.. MuSK frizzled-like domain is critical for mammalian neuromuscular junction formation and maintenance.. Journal of Neuroscience, 2015, 35 (12), pp.4926-4941. ⟨10.1523/JNEUROSCI.3381-14.2015⟩. ⟨hal-01165625⟩
- Sophie Nicole, Amina Chaouch, Torberg Torbergsen, Stéphanie Bauché, Elodie de Bruyckere, et al.. Agrin mutations lead to a congenital myasthenic syndrome with distal muscle weakness and atrophy. Brain - A Journal of Neurology , 2014, 137 (9), pp.2429-2443. ⟨10.1093/brain/awu160⟩. ⟨hal-03863959⟩
- Stéphanie Bauché, Delphine Boerio, Claire-Sophie Davoine, Véronique Bernard, Morgane Stum, et al.. Peripheral nerve hyperexcitability with preterminal nerve and neuromuscular junction remodeling is a hallmark of Schwartz-Jampel syndrome. Neuromuscular Disorders, 2013, 23 (12), pp.998-1009. ⟨10.1016/j.nmd.2013.07.005⟩. ⟨hal-03993872⟩
- Gaëlle Bruneteau, Thomas Simonet, Stéphanie Bauché, Nathalie Mandjee, Edoardo Malfatti, et al.. Muscle histone deacetylase 4 upregulation in amyotrophic lateral sclerosis: potential role in reinnervation ability and disease progression. Brain - A Journal of Neurology , 2013, 136 (8), pp.2359-2368. ⟨10.1093/brain/awt164⟩. ⟨hal-03993882⟩
- Asma Ben Ammar, Payam Soltanzadeh, Stéphanie Bauché, Pascale Richard, Evelyne Goillot, et al.. A Mutation Causes MuSK Reduced Sensitivity to Agrin and Congenital Myasthenia. PLoS ONE, 2013, 8 (1), pp.e53826. ⟨10.1371/journal.pone.0053826⟩. ⟨hal-01537840⟩
- I. Wargon, P. Richard, T. Kuntzer, D. Sternberg, S. Nafissi, et al.. Long-term follow-up of patients with congenital myasthenic syndrome caused by COLQ mutations. Neuromuscular Disorders, 2012, 22 (4), pp.318-324. ⟨10.1016/j.nmd.2011.09.002⟩. ⟨hal-03863780⟩
- Laure Strochlic, Julien Falk, Evelyne Goillot, Séverine Sigoillot, Francine Bourgeois, et al.. Wnt4 participates in the formation of vertebrate neuromuscular junction.. PLoS ONE, 2012, 7 (1), pp.e29976. ⟨10.1371/journal.pone.0029976⟩. ⟨hal-00720087⟩
- Karen Gaudon, Isabelle Pénisson-Besnier, Brigitte Chabrol, Françoise Bouhour, Laurence Demay, et al.. Multiexon deletions account for 15% of Congenital Myasthenic Syndrome with RAPSN mutations after negative DNA Sequencing. Journal of Medical Genetics, 2010, 47 (12), pp.795. ⟨10.1136/jmg.2010.081034⟩. ⟨hal-00574007⟩
- A. Ben Ammar, F. Petit, N. Alexandri, K. Gaudon, Stéphanie Bauché, et al.. Phenotype genotype analysis in 15 patients presenting a congenital myasthenic syndrome due to mutations in DOK7. Journal of Neurology, 2010, 257 (5), pp.754-766. ⟨10.1007/s00415-009-5405-y⟩. ⟨hal-03864208⟩
- Valérie Risson, Laetitia Mazelin, Mila Roceri, Hervé Sanchez, Vincent Moncollin, et al.. Muscle inactivation of mTOR causes metabolic and dystrophin defects leading to severe myopathy. Journal of Cell Biology, 2009, 187 (6), pp.859-874. ⟨10.1083/jcb.200903131⟩. ⟨hal-02126916⟩
- Caroline Huzé, Stéphanie Bauché, Pascale Richard, Frédéric Chevessier, Evelyne Goillot, et al.. Identification of an agrin mutation that causes congenital myasthenia and affects synapse function.. American Journal of Human Genetics, 2009, 85 (2), pp.155-67. ⟨10.1016/j.ajhg.2009.06.015⟩. ⟨inserm-00409064⟩
- P. Rigoard, K. Buffenoir, Stéphanie Bauché, J.-P. Giot, J. Koenig, et al.. Organisation structurale, moléculaire, formation et maturation de la jonction neuromusculaire. Neurochirurgie, 2009, 55, pp.S34-S42. ⟨10.1016/j.neuchi.2008.03.012⟩. ⟨hal-03863763⟩
- P. Rigoard, K. Buffenoir, M. Wager, Stéphanie Bauché, J.-P. Giot, et al.. Organisation anatomique et physiologique du nerf périphérique. Neurochirurgie, 2009, 55, pp.S3-S12. ⟨10.1016/j.neuchi.2008.03.009⟩. ⟨hal-03863732⟩
- S. Rigoard, M. Wager, K. Buffenoir, Stéphanie Bauché, J.-P. Giot, et al.. Principaux mécanismes impliqués dans la transmission synaptique au sein de l’appareil neuromusculaire. Neurochirurgie, 2009, 55, pp.S22-S33. ⟨10.1016/j.neuchi.2008.03.011⟩. ⟨hal-03863768⟩
- J. Koenig, Stéphanie Bauché, A. Ben Ammar, D. Nicolle, P. Rigoard, et al.. Remaniements expérimentaux et pathologiques de la jonction neuromusculaire. Neurochirurgie, 2009, 55, pp.S104-S109. ⟨10.1016/j.neuchi.2008.05.008⟩. ⟨hal-03863744⟩
- P. Rigoard, K. Buffenoir, M. Chaillou, M. Fares, L. da Costa, et al.. Existe-t-il un retentissement sur la jonction neuromusculaire de rat lors de lésions du système nerveux central ?. Neurochirurgie, 2009, 55, pp.S110-S123. ⟨10.1016/j.neuchi.2008.05.009⟩. ⟨hal-03863786⟩
- P. Rigoard, Stéphanie Bauché, K. Buffenoir, J.-P. Giot, J.-P. Faure, et al.. Le support anatomique de la contraction musculaire. Neurochirurgie, 2009, 55, pp.S69-S82. ⟨10.1016/j.neuchi.2008.05.006⟩. ⟨hal-03863717⟩
- P. Rigoard, K. Buffenoir, M. Wager, Stéphanie Bauché, J.-P. Giot, et al.. Architecture moléculaire du réticulum sarcoplasmique et son rôle dans le couplage excitation-contraction. Neurochirurgie, 2009, 55, pp.S83-S91. ⟨10.1016/j.neuchi.2008.05.007⟩. ⟨hal-03863756⟩
- S. D’houtaud, E. Sztermer, K. Buffenoir, J.-P. Giot, M. Wager, et al.. Formation et régénération synaptique. Neurochirurgie, 2009, 55, pp.S49-S62. ⟨10.1016/j.neuchi.2008.03.014⟩. ⟨hal-03863711⟩
- P. Rigoard, K. Buffenoir, Stéphanie Bauché, M. Fares, J. Koenig, et al.. Modalités et outils d’observation de la jonction neuromusculaire. Neurochirurgie, 2009, 55, pp.S43-S48. ⟨10.1016/j.neuchi.2008.03.013⟩. ⟨hal-03863752⟩
- P. Richard, K. Gaudon, H. Haddad, A. Ammar, E. Genin, et al.. The CHRNE 1293insG founder mutation is a frequent cause of congenital myasthenia in North Africa. Neurology, 2008, 71 (24), pp.1967-1972. ⟨10.1212/01.wnl.0000336921.51639.0b⟩. ⟨hal-03863833⟩
- Pascale Richard, Karen Gaudon, Emmanuel Fournier, Christopher Jackson, Stéphanie Bauché, et al.. A synonymous CHRNE mutation responsible for an aberrant splicing leading to congenital myasthenic syndrome. Neuromuscular Disorders, 2007, 17 (5), pp.409-414. ⟨10.1016/j.nmd.2007.01.018⟩. ⟨hal-03864196⟩
- Frédéric Chevessier, Stéphanie Bauché-Godard, Jean-Paul Leroy, Jeanine Koenig, Marion Paturneau-Jouas, et al.. The origin of tubular aggregates in human myopathies. Journal of Pathology, 2005, 207 (3), pp.313-323. ⟨10.1002/path.1832⟩. ⟨hal-03864293⟩
- Laure Strochlic, Annie Cartaud, Jean Cartaud. [14-3-3 gamma, a novel partner of MuSK downregulates synaptic gene transcription at the neuromuscular junction] Un nouveau partenaire de MuSK à la jonction neuromusculaire, la 14-3-3 gamma, réprime la transcription des gènes synaptiques. Médecine/Sciences, 2005, 21(5), pp.467-9. ⟨hal-00069250⟩
- Frédéric Chevessier, Brice Faraut, Aymeric Ravel-Chapuis, Pascale Richard, Karen Gaudon, et al.. Caractérisation physiopathologique des syndromes myasthéniques congénitaux : l'exemple de mutations dans le gène MUSK. Journal de la Société de Biologie, 2005, 199 (1), pp.61-77. ⟨10.1051/jbio:2005008⟩. ⟨hal-03864221⟩
- Laure Strochlic, Annie Cartaud, Jean Cartaud. The synaptic Muscle-Specific Kinase (MuSK) complex: new partners, new functions.. BioEssays, 2005, 27, pp.1129-35. ⟨10.1002/bies.20305⟩. ⟨hal-00008206v2⟩
- Laure Strochlic, Annie Cartaud, Alexandre Mejat, Régis Grailhe, Laurent Schaeffer, et al.. 14-3-3 gamma associates with muscle specific kinase and regulates synaptic gene transcription at vertebrate neuromuscular synapse.. Proceedings of the National Academy of Sciences of the United States of America, 2004, 101 (52), pp.18189-94. ⟨10.1073/pnas.0406905102⟩. ⟨pasteur-00163509⟩
- Frédéric Chevessier, Brice Faraut, Aymeric Ravel-Chapuis, Pascale Richard, Karen Gaudon, et al.. MUSK, a new target for mutations causing congenital myasthenic syndrome. Human Molecular Genetics, 2004, 13 (24), pp.3229-3240. ⟨10.1093/hmg/ddh333⟩. ⟨hal-03863846⟩
- B Mowry, P Holmans, A Pulver, P Gejman, B Riley, et al.. Multicenter linkage study of schizophrenia loci on chromosome 22q. Molecular Psychiatry, 2004, 9 (8), pp.784-795. ⟨10.1038/sj.mp.4001481⟩. ⟨hal-03863802⟩
- B. Eymard, C. Ioos, A. Barois, B. Estournet, M. Mayer, et al.. Syndromes myasthéniques congénitaux dus à des mutations du gène de la rapsyne. Revue Neurologique, 2004, 160 (5), pp.78-84. ⟨10.1016/s0035-3787(04)71009-7⟩. ⟨hal-03863776⟩
- Claudine Laurent, Dana Niehaus, Stéphanie Bauché, Douglas Levinson, Stéphane Soubigou, et al.. CAG repeat polymorphisms in KCNN3 (HSKCa3) and PPP2R2B show no association or linkage to schizophrenia. American Journal of Medical Genetics Part B: Neuropsychiatric Genetics, 2003, 116B (1), pp.45-50. ⟨10.1002/ajmg.b.10797⟩. ⟨hal-03863965⟩
- Douglas Levinson, Peter Holmans, Claudine Laurent, Brien Riley, Ann Pulver, et al.. No Major Schizophrenia Locus Detected on Chromosome 1q in a Large Multicenter Sample. Science, 2002, 296 (5568), pp.739-741. ⟨10.1126/science.1069914⟩. ⟨hal-03863693⟩
Conference papers
- A. Corvó, L. Matalonga, C. Hernandez-Ferrer, R. Thompson, L. Carmody, et al.. Treatabolome DB: linking gene and variants with treatments for rare diseases. Solve-RD, Solving the unsolved Rare Diseases Final Meeting 2023, Apr 2023, Prague, Czech Republic. ⟨hal-04086225⟩
- Piera Smeriglio. Deregulation of TET/5hmC axis in motor neuron diseases. Epigenetic in muscle disease workshop, Jun 2022, PARIS, France. ⟨hal-04002798⟩
- Charles Frison-Roche, Steve Cottin, Jeanne Lainé, Ludovic Arandel, Marius Halliez, et al.. MBNL loss of function in the motor unit alters neuromuscular communication. International Myotonic Dystrophy Consortium Meeting IDMC-13, International Myotonic Dystrophy Consortium, May 2022, Osaka, Japan. ⟨hal-04005805⟩
- Strochlic Laure. Wnt signaling in Myasthenia. Guarda meeting on the neuromuscular junction, 2022, Guarda, Switzerland. ⟨hal-04002946⟩
- Gaëlle Bruneteau. Réseau ACT4ALS-MND - Actualités et perspectives - Essais thérapeutiques. Journées nationales annuelles de la filière de santé maladies rares FILSLAN, 2022, Montpellier, France. ⟨hal-04015822⟩
- Charles Frison-Roche, Julien Messéant, Jeanne Lainé, Ludovic Arandel, Mégane Lemaitre, et al.. MBNL loss of function in motoneurons leads to motor unit dysfunction in myotonic dystrophy. Journées de la Société Française de Myologie, Société Française de Myologie, Nov 2021, Saint-Etienne, France. ⟨hal-03999311⟩
- A Atalaia, C Hernandez Ferrer, A Corvó, L Matalonga, R Thompson, et al.. The Treatabolome flags treatable genes and variants: an emerging concept. ERN Euro-NMD Webinar on Treatabolome, Oct 2021, Webinar, Germany. ⟨hal-03989142⟩
- Gaëlle Bruneteau. Essais thérapeutiques en cours : consortium ACT4ALS-MND. Journées nationales annuelles de la filière de santé maladies rares FILSLAN-eJNA, 2021, Par visioconférence, France. ⟨hal-04015835⟩
- Strochlic Laure. GSK3-beta inhibition for therapy of CMS. 35th ENMC International Workshop on Congenital myasthenic syndromes, 2021, Par visioconférence, Netherlands. ⟨hal-04015990⟩
- Gaëlle Bruneteau. Réseau national de recherche clinique dans la SLA et les maladies du motoneurone. Journées nationales annuelles de la filière de santé maladies rares FILSLAN – eJNA, 2020, Par visioconférence, France. ⟨hal-04015843⟩
- Stéphanie Bauché. Mecanismes physiopathologiques impliqués dans les syndromes myasthéniques congénitaux. journée de Neuropédiatrie de Trousseau, Apr 2019, Paris, France. ⟨hal-03994234⟩
- Strochlic Laure. Regulation of neuromuscular connectivity by Wnt signaling. 6th International Congress of Myology, France, 2019, Bordeaux (France), France. ⟨hal-04002976⟩
- Strochlic Laure. Regulation of neuromuscular connectivity by Wnt signaling. Regulation of neuromuscular connectivEMBO Workshop on Molecular mechanisms of muscle wasting during aging and disease, 2018, Ascona, Switzerland. ⟨hal-04002986⟩
- Stéphanie Bauché, Geoffroy Vellieux, Damien Sternberg, Marie-Joséphine Fontenille, Guy Brochier, et al.. Mutations in GFPT1-related congenital myasthenic syndromes are associated with synaptic morphological defects and underlie a tubular aggregates myopathy with synaptopathy. journées de la société française de myologie, Nov 2017, Colmar, France. ⟨hal-03994005⟩
Poster communications
- France Piétri‐rouxel, Aly BOURGUIBA VILLENEUVE, Sestina Falcone, Sonia Pezet, Massiré Traoré, et al.. Unraveling the role of GDF5 therapeutic potential in Amyotrophic Lateral Sclerosis. Myology 2024, Apr 2024, Paris, France. ⟨hal-04782464⟩
- Stéphanie Bauché, Marion Masingue, Olivia Cattaneo, Damien Sternberg, Céline Buon, et al.. New mutation in the beta 1 propeller domain of LRP4 responsible for congenital myasthenic syndrome associated with Cenani-Lenz syndrome. journées de la société française de myologie, Nov 2022, Toulouse, France. ⟨hal-03994163⟩
- Sestina Falcone, Marais T., Traoré M., Bourguiba A., Gentil C., et al.. Unraveling the role of GDF5 therapeutic potential in Amyotrophic Lateral Sclerosis. 19 Journée de la societé Française de Myologie, Nov 2022, Toulouse (FR), France. ⟨hal-04002173⟩
- Charles Frison-Roche, Steve Cottin, Jeanne Lainé, Ludovic Arandel, Marius Halliez, et al.. MBNL loss of function in motoneurons leads to motor unit dysfunction in myotonic dystrophy. 7th International Myology Congress, Sep 2022, Nice, France. ⟨hal-04005838⟩
- Sestina Falcone, T. Marais, M. Traoré, C. Gentil, J. Mésseant, et al.. Unraveling the role of GDF5 therapeutic potential in Amyotrophic Lateral Sclerosis. Myology 2022, Sep 2022, Nice (FRANCE), France. ⟨hal-04002164⟩
- Massiré Traoré, Chiara Noviello, Gentil Christel, Julien Messéant, Ericky Caldas, et al.. Therapeutic approach based on GDF5 to counteract age-related muscle wasting. Muscle formation, maintenance, regeneration and pathology-EMBO workshop, Apr 2022, Gouvieux (FR), France. ⟨hal-04002159⟩
- Halliez Marius, Cottin Steve, You Axel, Lemaitre Mégane, Ezan Jérome, et al.. MUSK CYSTEIN RICH DOMAIN IN AUTOIMMUNE MYASTHENIC SYNDROME. 7th International Myology Congress, 2022, Nice, France. ⟨hal-04002800⟩
- Halliez Marius, Cottin Steve, You Axel, Lemaitre Mégane, Ezan Jérome, et al.. MUSK CYSTEIN RICH DOMAIN IN AUTOIMMUNE MYASTHENIC SYNDROME. 14th MGFA International Conference On Myasthenia And Related Disorders, 2022, Miami (Florida), United States. ⟨hal-04002779⟩
- Sestina Falcone, T. Marais, M. Traoré, C. Gentil, J. Mésseant, et al.. Unraveling the role of GDF5 therapeutic potential in Amyotrophic Lateral Sclerosis. 18ème Journée de la Societé Française de Myologie, Nov 2021, Saint-Etienne (FR), France. ⟨hal-04002180⟩
- Stéphanie Bauché, Damien Sternberg, Céline Buon, Julien Messéant, Myriam Boëx, et al.. Identification of a new splice site mutation in synaptotagmin-2 responsible for a severe and early presynaptic form of congenital myasthenic syndrome. World muscle society, Sep 2021, Online, France. ⟨hal-03994061⟩
- Carles Hernandez-Ferrer, Leslie Matalonga, Rachel Thompson, Leigh Carmody, Davide Piscia, et al.. The Treatabolome Database and Platform: enhancing Rare Diseases’ treatment visibility.. European Human Genetics Virtual Conference 2021, Aug 2021, Virtual conference, United Kingdom. ⟨hal-03988844⟩
- G Bruneteau, D Devos, A Bordet, E Bernard, Jp Camdessanche, et al.. A French national network to improve organization and inclusion in clinical trials: Alliance on Clinical Trials for ALS-MND (ACT4ALS-MND). ALS-MND congress, 2021, Par visioconférence, France. ⟨hal-04015853⟩
- A Hesters, Mdm Amador, R Debs, N Le Forestier, T Lenglet, et al.. Facteurs pronostiques après pose de gastrostomie chez les patients atteints de SLA utilisateurs habituels de VNI : influence du statut respiratoire. Journées Recherche FilSLAN/ARSLA (e-JR6 SLA), 2020, Par visioconférence, France. ⟨hal-04015862⟩
- Stéphanie Bauché, Sureau Alain, Damien Sternberg, Céline Buon, Julien Messeant, et al.. Identification of a new splice site mutation in synaptotagmin-2 responsible for a severe and early presynaptic form of congenital myasthenic syndrome. journées de la société française de myologie, Nov 2019, Marseille (13), France. ⟨hal-03994043⟩
- Maximilien Sochala, Raphaël Porcher, Tanya Stojkovic, Henri Marc Bécane, Anthony Béhin, et al.. High Risk of Fatal and Non-Fatal Venous Thromboembolism in Myotonic Dystrophy. 6th International Congress of Myology, Mar 2019, Bordeaux, France. ⟨hal-04020135⟩
- Stéphanie Bauché, Damien Sternberg, Julien Messéant, Myriam Boëx, Marie-Christine Nouguès, et al.. Recent advances in french cohort of congenital myasthenic syndromes patients. Myology, Mar 2019, Bordeaux, France. ⟨hal-03994184⟩
- J Messéant, M Boex, S Bauché, A Dobbertin, C Legay, et al.. Wnt/MuSK/ß-catenin signaling: a complex regulator of neuromuscular connectivity. 6th International Congress of Myology, France, 2019, Bordeaux (France), France. ⟨hal-04002695⟩
- R Guimarães-Costa, Mc Niérat, I Rivals, C Morélot-Panzini, Nb Romero, et al.. Diaphragm pacing impairs local myofiber reinnervation in Amyotrophic Lateral Sclerosis. ALS-MND congress, 2019, Perth (Australia), Australia. ⟨hal-04015875⟩
- Myriam Boex, Julien Messéant, Stéphanie Bauché, Marius. Halliez, Bertrand Fontaine, et al.. A new Vangl2/MuSK signaling pathway promotes neuromuscular junction formation by enhancing postsynaptic assembly and motor axon growth stop. 6th International Congress of Myology, France, 2019, Bordeaux (France), France. ⟨hal-04002674⟩
- Stéphanie Bauché, Geoffroy Vellieux, Damien Sternberg, Michel Fardeau, Emmanuelle Lacène, et al.. Mutations in GFPT1-related congenital myasthenic syndromes underlie a tubular aggregates myopathy with synaptopathy. international congress of neuromuscular diseases, Jul 2018, Vienna (Austria), Austria. ⟨hal-03994257⟩
- M Boex, J Messéant, S Bauché, B Fontaine, L Strochlic. Role of Vangl2, a core component of the Wnt Planar cell Polarity pathway during neuromuscular junction formation. 15th International Congress on Neuromuscular Diseases, 2018, Vienne (Autriche), Austria. ⟨hal-04002728⟩
Theses
- Marius Halliez. Le domaine riche en cystéine de MuSK dans la myasthénie auto-immune. Biologie cellulaire. Sorbonne Université, 2023. Français. ⟨NNT : 2023SORUS093⟩. ⟨tel-04137804⟩
- Myriam Boëx. Role of MuSK/Vangl2 signaling pathway in neuromuscular connectivity. Cellular Biology. Sorbonne Université, 2020. English. ⟨NNT : 2020SORUS258⟩. ⟨tel-03787911⟩
Powered by HAL ![]()


