

Repeat Expansions & Myotonic Dystrophy (REDs)
La thématique principale de l’équipe est centrée sur la Dystrophie Myotonique, une des maladies neuromusculaires les plus fréquentes chez l’adulte, et plus particulièrement sur la DM de type 1 (DM1) également appelée maladie de Steinert. La DM1 est caractérisée par une faiblesse et une atrophie musculaire progressive, une myotonie, des défauts de conduction cardiaque, une cataracte précoce, des troubles endocriniens et gastro-intestinaux ainsi que des atteintes neurologiques. On distingue cinq formes cliniques de cette maladie multisystémique incluant les formes tardive, adulte, juvénile, infantile et congénitale. Actuellement, il n’y a pas de traitement pour cette maladie génétique mais diverses approches thérapeutiques sont en développement.
La DM1 est une maladie autosomique dominante causée par une expansion anormale de triplets CTGn (n>40) localisée dans la région 3’ non codante du gène DMPK. Le caractère instable de cette expansion de séquences CTG répétées se manifeste dans différents tissus tout au long de la vie du patient mais également entre générations successives, et représente la base moléculaire du phénomène d’anticipation observé dans cette maladie. La DM1 est une maladie à gain-de-fonction d’ARN. En effet, les transcrits DMPK mutés contenant des répétitions CUG pathologiques (ARN-CUGexp) sous retenus dans le noyau des cellules sous forme d’agrégats riboprotéiques (foci) et perturbent les fonctions de certaines protéines de liaison aux ARNs (RBPs). Notamment les foci séquestrent les RBPs de la famille MBNL impliquées dans le processus de maturation des ARNs. Ainsi, la perte fonctionnelle de MBNL conduit à des défauts d’épissage alternatif de certains ARN pré-messagers qui ont été associés à des symptômes comme ceux du CLCN1 à la myotonie, de INSR à la résistance à l’insuline, de BIN1 à la faiblesse musculaire, de DMD à l’altération de l’architecture des fibres musculaires et de SCN5A à des défauts de conduction et du rythme cardiaque. Cependant des mécanismes additionnels sont impliqués dans les processus physiopathologiques complexes de cette maladie qui affecte divers types cellulaires et de nombreux tissus.
L’équipe REDs a été créée en 2019 suite à la fusion des équipes de Geneviève Gourdon et de Denis Furling. Elle inclut également le groupe de Guillaume Bassez, neurologue qui coordonne le registre national de dystrophie myotonique (DM-Scope) et celui d’Arnaud Ferry qui s’intéresse à la physiologie musculaire et à l’exercice. L’objectif de cette nouvelle équipe est de synergiser les efforts afin d’accélérer la recherche translationnelle pour cette maladie neuromusculaire et de faire émerger des alternatives thérapeutiques pour les patients. Ainsi les compétences de l’équipe visent à réaliser une recherche intégrée autour de la DM1 allant des mécanismes fondamentaux de l’instabilité des expansions CTG, à la compréhension des mécanismes physiopathologiques à l’aide de modèles cellulaires et animaux, au développement et à l’évaluation d’approches thérapeutiques innovantes et enfin, à la mise en place d’essais pré-cliniques et cliniques pour cette maladie neuromusculaire.
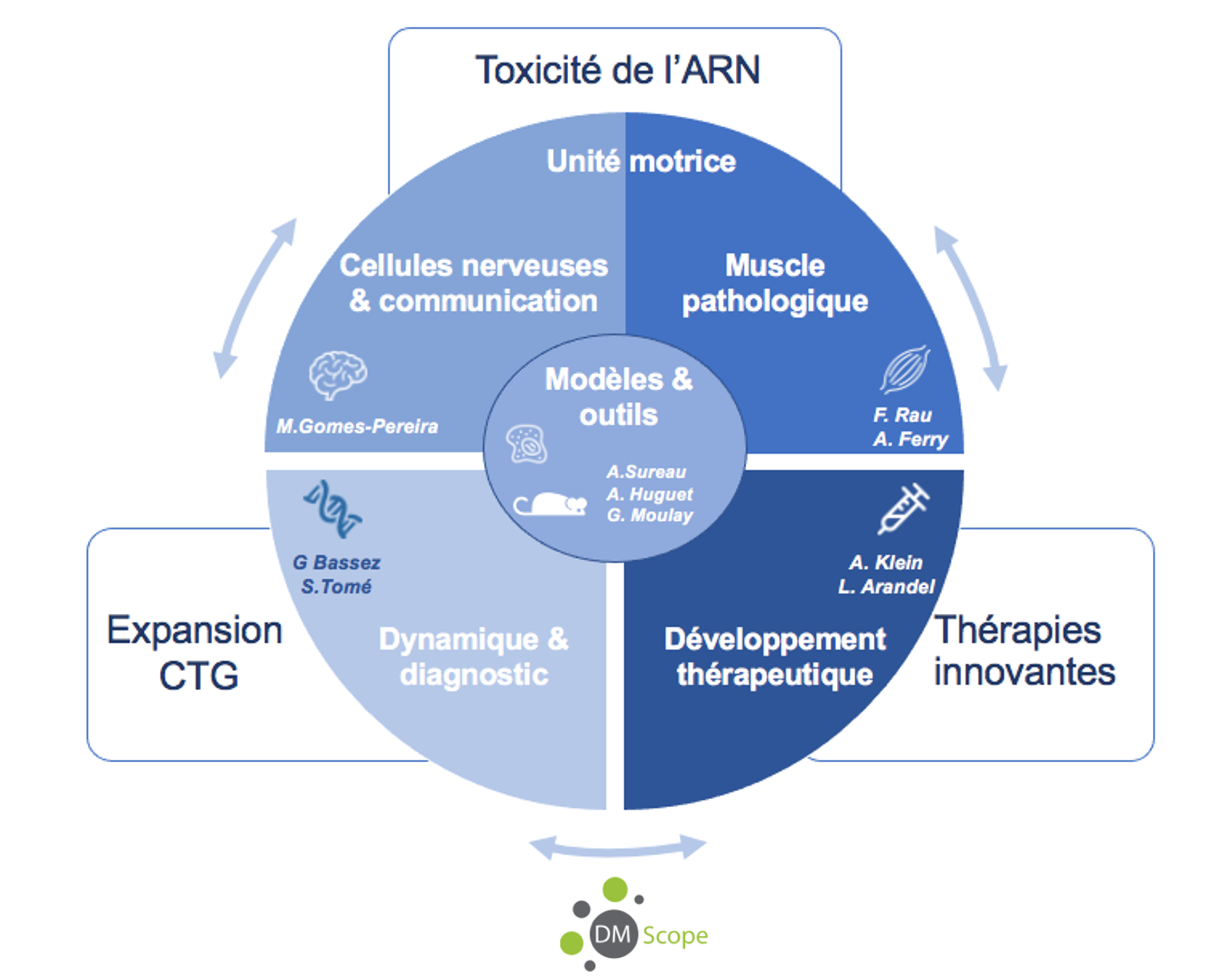
Thèmes de recherche
Dynamique des expansions CTG et implication pour le diagnostic (PIs-G. Bassez & S. Tomé)
A partir de modèle murins et cellulaires nous étudions les mécanismes d’instabilité des répétitions CTG et les facteurs permettant une modulation de leur dynamique. En parallèle, nous étudions des familles DM1 atypiques en lien avec le registre de patients DM-Scope et le service de neuromyologie. Les données croisées entre le laboratoire et la clinique permettrons d’affiner les outils diagnostique et pronostique.
Toxicité de l’ARN et conséquences physiopathologiques
Nous étudions l’impact de la mutation et de l’expression des ARN porteurs d’amplification (ARN-CUGexp) suivant un axe système nerveux/muscle et en particulier sur :
- Les conséquences moléculaires et fonctionnelles sur les différents types de cellules nerveuses et leur communication (PI-M. Gomes Pereira)
- Les motoneurones, l’unité motrice et la fonction musculaire (PIs- F. Rau & A Ferry)
Développement de nouvelles thérapies (PIs : A. Klein & L. Arandel)
Notre équipe développe diverses approches thérapeutiques innovantes pour la DM1, visant différents organes (muscle, cerveau, cœur..) testés à l’aide de modèles cellulaires et murins avec pour objectif le transfert direct vers les services cliniques de l’institut.
Modèles et outils (PIs : A. Surreau , A. Huguet-Lachon & G. Moulay)
De nouveaux modèles cellulaires, murins et outils moléculaires sont développés parallèlement à l’évolution des différents projets.
DM-Scope et recherche clinique (PI: G. Bassez)
- Coordination de l’Observatoire Français des Dystrophies Myotoniques – DM-Scope –
- Caractérisation phénotypique et de l’histoire naturelle de la Dystrophie Myotonique
- Validation d’outils de mesure pour les essais cliniques
- Essais cliniques



Contacts :
| Nom | Position | ORCID |
|---|


Articles dans une revue
- Baptiste Bogard, Hélène Bonnet, Ekaterina Boyarchuk, Gilles Tellier, Denis Furling, et al.. Small nucleolar RNAs promote the restoration of muscle differentiation defects in cells from myotonic dystrophy type 1. Nucleic Acids Research, 2025, 53 (6), ⟨10.1093/nar/gkaf232⟩. ⟨hal-05057978⟩
- Charles Frison-Roche, Célia Martin Demier, Steve Cottin, Jeanne Lainé, Ludovic Arandel, et al.. MBNL deficiency in motor neurons disrupts neuromuscular junction maintenance and gait coordination. Brain - A Journal of Neurology , 2025, 148 (4), pp.1180-1193. ⟨10.1093/brain/awae336⟩. ⟨hal-05045240⟩
- Dylan Moutachi, Janek Hyzewicz, Pauline Roy, Mégane Lemaitre, Damien Bachasson, et al.. Treadmill running and mechanical overloading improved the strength of the plantaris muscle in the dystrophin‐desmin double knockout (DKO) mouse. The Journal of Physiology, In press, ⟨10.1113/JP286425⟩. ⟨hal-04643936⟩
- Medhi Hassani, Dylan Moutachi, Mégane Lemaitre, Alexis Boulinguiez, Denis Furling, et al.. Beneficial effects of resistance training on both mild and severe mouse dystrophic muscle function as a preclinical option for Duchenne muscular dystrophy. PLoS ONE, 2024, 19, ⟨10.1371/journal.pone.0295700⟩. ⟨hal-04501283⟩
- Antonio Atalaia, Dagmar Wandrei, Nawel Lalout, Rachel Thompson, Adrian Tassoni, et al.. EURO-NMD registry: federated FAIR infrastructure, innovative technologies and concepts of a patient-centred registry for rare neuromuscular disorders. Orphanet Journal of Rare Diseases, 2024, 19 (1), pp.66. ⟨10.1186/s13023-024-03059-3⟩. ⟨hal-04460667⟩
- Anita Kneppers, Sabrina Ben Larbi, Marine Theret, Audrey Saugues, Carole Dabadie, et al.. AMPKα2 is a skeletal muscle stem cell intrinsic regulator of myonuclear accretion. iScience, 2023, 26 (12), pp.108343. ⟨10.1016/j.isci.2023.108343⟩. ⟨hal-04729009⟩
- Maggie Lutz, Miranda Levanti, Rebekah Karns, Genevieve Gourdon, Diana Lindquist, et al.. Therapeutic Targeting of the GSK3β-CUGBP1 Pathway in Myotonic Dystrophy. International Journal of Molecular Sciences, 2023, 24 (13), pp.10650. ⟨10.3390/ijms241310650⟩. ⟨hal-04259888⟩
- Florent Porquet, Lin Weidong, Kévin Jehasse, Hélène Gazon, Maria Kondili, et al.. Specific DMPK-promoter targeting by CRISPRi reverses myotonic dystrophy type 1-associated defects in patient muscle cells. Molecular Therapy - Nucleic Acids, 2023, 32, pp.857 - 871. ⟨10.1016/j.omtn.2023.05.007⟩. ⟨hal-04287597⟩
- Julie Tahraoui-Bories, Antoine Mérien, Anchel González-Barriga, Jeanne Lainé, Céline Leteur, et al.. MBNL‐dependent impaired development within the neuromuscular system in myotonic dystrophy type 1. Neuropathology and Applied Neurobiology, 2023, 49 (1), ⟨10.1111/nan.12876⟩. ⟨hal-03992575⟩
- Dylan Moutachi, Mégane Lemaitre, Clément Delacroix, Onnik Agbulut, Denis Furling, et al.. Valproic acid reduces muscle susceptibility to contraction‐induced functional loss but increases weakness in two murine models of Duchenne muscular dystrophy. Clinical and Experimental Pharmacology and Physiology, In press, ⟨10.1111/1440-1681.13804⟩. ⟨hal-04146953⟩
- Tanya Stojkovic, Marion Masingue, Helène Turmel, Marianne Hezode-Arzel, Anthony Béhin, et al.. Diagnostic yield of a practical electrodiagnostic protocol discriminating between different congenital myasthenic syndromes. Neuromuscular Disorders, 2022, 32 (11-12), pp.870-878. ⟨10.1016/j.nmd.2022.10.001⟩. ⟨hal-04074000⟩
- Alexandra Monceau, Dylan Moutachi, Mégane Lemaitre, Luis Garcia, Capucine Trollet, et al.. Dystrophin Restoration after Adeno-Associated Virus U7–Mediated Dmd Exon Skipping Is Modulated by Muscular Exercise in the Severe D2-Mdx Duchenne Muscular Dystrophy Murine Model. American Journal of Pathology, 2022, 192 (11), pp.1604-1618. ⟨10.1016/j.ajpath.2022.07.016⟩. ⟨hal-04308024⟩
- Yu-Chih Tsai, Laure de Pontual, Cheryl Heiner, Tanya Stojkovic, Denis Furling, et al.. Identification of a CCG-enriched expanded allele in patients with myotonic dystrophy type 1 using amplification-free long-read sequencing. Journal of Molecular Diagnostics, In press, ⟨10.1016/j.jmoldx.2022.08.003⟩. ⟨hal-03832574⟩
- Alexandra Monceau, Dylan Moutachi, Mégane Lemaitre, Luis Garcia, Capucine Trollet, et al.. Dystrophin Restoration after Adeno-Associated Virus U7–Mediated Dmd Exon Skipping Is Modulated by Muscular Exercise in the Severe D2-Mdx Duchenne Muscular Dystrophy Murine Model. American Journal of Pathology, 2022, ⟨10.1016/j.ajpath.2022.07.016⟩. ⟨hal-03830848⟩
- Siham Ait Benichou, Dominic Jauvin, Thiéry de Serres-Bérard, Frank Bennett, Frank Rigo, et al.. Enhanced Delivery of Ligand-Conjugated Antisense Oligonucleotides (C16-HA-ASO) Targeting Dystrophia Myotonica Protein Kinase Transcripts for the Treatment of Myotonic Dystrophy Type 1. Human Gene Therapy, 2022, 33 (15-16), pp.810-820. ⟨10.1089/hum.2022.069⟩. ⟨hal-03753318⟩
- Myriam Boëx, Steve Cottin, Marius Halliez, Stéphanie Bauché, Céline Buon, et al.. The cell polarity protein Vangl2 in the muscle shapes the neuromuscular synapse by binding to and regulating the tyrosine kinase MuSK. Science Signaling, 2022, 15 (734), pp.eabg4982. ⟨10.1126/scisignal.abg4982⟩. ⟨inserm-03768653⟩
- Benoît Sanson, Caroline Stalens, Céline Guien, Luisa Villa, Catherine Eng, et al.. Convergence of patient- and physician-reported outcomes in the French National Registry of Facioscapulohumeral Dystrophy. Orphanet Journal of Rare Diseases, 2022, 17 (1), pp.96. ⟨10.1186/s13023-021-01793-6⟩. ⟨hal-03596523⟩
- Beatrice Cardinali, Claudia Provenzano, Mariapaola Izzo, Christine Voellenkle, Jonathan Battistini, et al.. Time-controlled and muscle-specific CRISPR/Cas9-mediated deletion of CTG-repeat expansion in the DMPK gene. Molecular Therapy - Nucleic Acids, 2022, 27, pp.184-199. ⟨10.1016/j.omtn.2021.11.024⟩. ⟨hal-03511841⟩
- Ludovic Arandel, Magdalena Matloka, Arnaud F Klein, Frédérique Rau, Alain Sureau, et al.. Reversal of RNA toxicity in myotonic dystrophy via a decoy RNA-binding protein with high affinity for expanded CUG repeats. Nature Biomedical Engineering, 2022, 6 (2), pp.207-220. ⟨10.1038/s41551-021-00838-2⟩. ⟨hal-03830811⟩
- Siham Ait Benichou, Dominic Jauvin, Thiéry de Serres-Bérard, Marion Pierre, Karen K Ling, et al.. Antisense oligonucleotides as a potential treatment for brain deficits observed in myotonic dystrophy type 1. Gene Therapy, 2022, ⟨10.1038/s41434-022-00316-7⟩. ⟨hal-03544255⟩
- Brigitte Potier, Louison Lallemant, Sandrine Parrot, Aline Huguet-Lachon, Geneviève Gourdon, et al.. DM1 Transgenic Mice Exhibit Abnormal Neurotransmitter Homeostasis and Synaptic Plasticity in Association with RNA Foci and Mis-Splicing in the Hippocampus. International Journal of Molecular Sciences, 2022, 23 (2), pp.592. ⟨10.3390/ijms23020592⟩. ⟨hal-03542361⟩
- Remco T P van Cruchten, Daniël van As, Jeffrey C Glennon, Baziel G M van Engelen, K Okkersen, et al.. Clinical improvement of DM1 patients reflected by reversal of disease-induced gene expression in blood. BMC Medicine, 2022, 20 (1), pp.395. ⟨10.1186/s12916-022-02591-y⟩. ⟨hal-03848010⟩
- Diana Dincã, Louison Lallemant, Anchel González-Barriga, Noémie Cresto, Sandra Braz, et al.. Myotonic dystrophy RNA toxicity alters morphology, adhesion and migration of mouse and human astrocytes. Nature Communications, 2022, 13 (1), pp.3841. ⟨10.1038/s41467-022-31594-9⟩. ⟨hal-03715682⟩
- Abdallah Fayssoil, Lee S Nguyen, Tanya Stojkovic, Helene Prigent, Robert Carlier, et al.. Determinants of diaphragm inspiratory motion, diaphragm thickening, and its performance for predicting respiratory restrictive pattern in Duchenne muscular dystrophy. Muscle & Nerve, 2022, 65 (1), pp.89-95. ⟨10.1002/mus.27432⟩. ⟨hal-03521139⟩
- Antoine Mérien, Julie Tahraoui-Bories, Michel Cailleret, Jean-Baptiste Dupont, Céline Leteur, et al.. CRISPR gene editing in pluripotent stem cells reveals the function of MBNL proteins during human in vitro myogenesis. Human Molecular Genetics, 2022, 31 (1), pp.41-56. ⟨10.1093/hmg/ddab218⟩. ⟨hal-03830948⟩
- Sandrine Parrot, Alex Corscadden, Louison Lallemant, Hélène Benyamine, Jean-Christophe Comte, et al.. Defects in Mouse Cortical Glutamate Uptake Can Be Unveiled In Vivo by a Two-in-One Quantitative Microdialysis. ACS Chemical Neuroscience, 2022, 13 (1), pp.134 - 142. ⟨10.1021/acschemneuro.1c00634⟩. ⟨hal-03753548⟩
- Alexandra Monceau, Clément Delacroix, Mégane Lemaitre, Gaelle Revet, Denis Furling, et al.. The beneficial effect of chronic muscular exercise on muscle fragility is increased by Prox1 gene transfer in dystrophic mdx muscle. PLoS ONE, 2022, 17 (4), pp.e0254274. ⟨10.1371/journal.pone.0254274⟩. ⟨hal-03830867⟩
- Laure de Pontual, Stéphanie Tomé. Overview of the Complex Relationship between Epigenetics Markers, CTG Repeat Instability and Symptoms in Myotonic Dystrophy Type 1. International Journal of Molecular Sciences, 2022, 23 (7), pp.3477. ⟨10.3390/ijms23073477⟩. ⟨hal-03832569⟩
- Cécile Delorme, Marion Houot, Charlotte Rosso, Stéphanie Carvalho, Thomas Nedelec, et al.. The wide spectrum of COVID-19 neuropsychiatric complications within a multidisciplinary centre. Brain Communications, 2021, 3 (3), ⟨10.1093/braincomms/fcab135⟩. ⟨hal-03464402⟩
- Gilles Moulay, Marc Bitoun, Denis Furling, Steṕhane Vassilopoulos. Comment l’épissage alternatif contribue au contrôle de la plasticité des structures de clathrine. Médecine/Sciences, 2021, 37 (12), pp.1186-1188. ⟨10.1051/medsci/2021178⟩. ⟨hal-03498232⟩
- Shira Yanovsky-Dagan, Eliora Cohen, Pauline Megalli, Gheona Altarescu, Oshrat Schonberger, et al.. DMPK hypermethylation in sperm cells of myotonic dystrophy type 1 patients. European Journal of Human Genetics, In press, ⟨10.1038/s41431-021-00999-3⟩. ⟨hal-03449010⟩
- Laure de Pontual, Geneviève Gourdon, Stéphanie Tomé. Identification of new factors inducing CTG.CAG repeat contractions in Myotonic Dystrophy type 1. Médecine/Sciences, 2021, 37, pp.6 - 10. ⟨10.1051/medsci/2021182⟩. ⟨hal-03753522⟩
- Maryvonne Ardourel, Chloé Felgerolle, Arnaud Pâris, Niyazi Acar, Khaoula Ramchani Ben Othman, et al.. Dietary supplement enriched in antioxidants and omega-3 promotes glutamine synthesis in Müller cells: a key process against oxidative stress in retina. Nutrients, 2021, 13 (9), pp.3216. ⟨10.3390/nu13093216⟩. ⟨hal-03356546⟩
- S Birnbaum, R Porcher, P Portero, B Clair, S Demeret, et al.. Home-based exercise in autoimmune myasthenia gravis: A randomized controlled trial. Neuromuscular Disorders, 2021, 31 (8), pp.726-735. ⟨10.1016/j.nmd.2021.05.002⟩. ⟨hal-03442076⟩
- Daniël van As, Kees Okkersen, Guillaume Bassez, Benedikt Schoser, Hanns Lochmüller, et al.. Clinical Outcome Evaluations and CBT Response Prediction in Myotonic Dystrophy. Journal of Neuromuscular Diseases, 2021, 8 (6), pp.1031 - 1046. ⟨10.3233/jnd-210634⟩. ⟨hal-03463099⟩
- Aurélie Kas, Marine Soret, Nadya Pyatigoskaya, Marie-Odile Habert, Adèle Hesters, et al.. The cerebral network of COVID-19-related encephalopathy: a longitudinal voxel-based 18F-FDG-PET study. European Journal of Nuclear Medicine and Molecular Imaging, 2021, 48 (8), pp.2543-2557. ⟨10.1007/s00259-020-05178-y⟩. ⟨hal-03769581⟩
- Daniela Rovito, Anna-Isavella Rerra, Vanessa Ueberschlag-Pitiot, Shilpy Joshi, Nezih Karasu, et al.. Myod1 and GR coordinate myofiber-specific transcriptional enhancers. Nucleic Acids Research, 2021, 49 (8), pp.4472 - 4492. ⟨10.1093/nar/gkab226⟩. ⟨hal-03251776⟩
- Gorka Fernández-Eulate, Giorgia Querin, Ursula Moore, Anthony Behin, Marion Masingue, et al.. Deep phenotyping of an international series of patients with late‐onset dysferlinopathy. European Journal of Neurology, 2021, 28 (6), pp.2092-2102. ⟨10.1111/ene.14821⟩. ⟨hal-03263341⟩
- Raphaël Porcher, Isabelle Desguerre, Helge Amthor, Brigitte Chabrol, Frédérique Audic, et al.. Association between prophylactic angiotensin-converting enzyme inhibitors and overall survival in Duchenne muscular dystrophy—analysis of registry data. European Heart Journal, 2021, ⟨10.1093/eurheartj/ehab054⟩. ⟨hal-03179750⟩
- Anchel González-Barriga, Louison Lallemant, Diana M Dincã, Sandra O Braz, Hélène Polvèche, et al.. Integrative Cell Type-Specific Multi-Omics Approaches Reveal Impaired Programs of Glial Cell Differentiation in Mouse Culture Models of DM1. Frontiers in Cellular Neuroscience, 2021, 15, pp.662035. ⟨10.3389/fncel.2021.662035⟩. ⟨hal-03235993⟩
- Caroline Stalens, Leslie Motté, Anthony Béhin, Rabah Ben Yaou, France Leturcq, et al.. Improved cardiac outcomes by early treatment with angiotensin-converting enzyme inhibitors in Becker muscular dystrophy. Journal of Neuromuscular Diseases, 2021, 8 (4), pp.495 - 502. ⟨10.3233/jnd-200620⟩. ⟨hal-03464423⟩
- Stephan Wenninger, Sarah Cumming, Kristina Gutschmidt, Kees Okkersen, Aura Cecilia Jimenez-Moreno, et al.. Associations Between Variant Repeat Interruptions and Clinical Outcomes in Myotonic Dystrophy Type 1. Neurology Genetics, 2021, 7 (2), pp.e572. ⟨10.1212/NXG.0000000000000572⟩. ⟨hal-03875210⟩
- Valentina Grande, Denisa Hathazi, Emily O’connor, Theo Marteau, Ulrike Schara-Schmidt, et al.. Dysregulation of GSK3β-Target Proteins in Skin Fibroblasts of Myotonic Dystrophy Type 1 (DM1) Patients. Journal of Neuromuscular Diseases, 2021, 8, pp.603 - 619. ⟨10.3233/jnd-200558⟩. ⟨hal-03463068⟩
- Virginie Lambrecq, Aurélie Hanin, Esteban Munoz-Musat, Lydia Chougar, Salimata Gassama, et al.. Association of clinical, biological, and brain magnetic resonance imaging findings with electroencephalographic findings for patients with COVID-19. JAMA Network Open, 2021, 4 (3), pp.e211489. ⟨10.1001/jamanetworkopen.2021.1489⟩. ⟨hal-04015178⟩
- Mário Gomes-Pereira, Darren G Monckton. Chronic Exposure to Cadmium and Antioxidants Does Not Affect the Dynamics of Expanded CAG•CTG Trinucleotide Repeats in a Mouse Cell Culture System of Unstable DNA. Frontiers in Cellular Neuroscience, 2021, 14, pp.606331. ⟨10.3389/fncel.2020.606331⟩. ⟨hal-03148527⟩
- Denis Furling. Cas9 targeting of toxic foci of RNA repeats. Nature Biomedical Engineering, 2021, 5 (2), pp.130-131. ⟨10.1038/s41551-021-00688-y⟩. ⟨hal-03141957⟩
- Narjes Baati, Nathalie Mougenot, Mégane Lemaitre, Marine Kirsch, Onnik Agbulut, et al.. Alteration of skeletal and cardiac muscles function in DBA/2J mdx mice background: a focus on high intensity interval training. Intractable & Rare Diseases Research, 2021, 10 (4), pp.269-275. ⟨10.5582/irdr.2021.01097⟩. ⟨hal-03830933⟩
- Antoine Mangin, Laure de Pontual, Yu-Chih Tsai, Laetitia Monteil, Mathilde Nizon, et al.. Robust Detection of Somatic Mosaicism and Repeat Interruptions by Long-Read Targeted Sequencing in Myotonic Dystrophy Type 1. International Journal of Molecular Sciences, 2021, 22 (5), pp.2616. ⟨10.3390/ijms22052616⟩. ⟨hal-03173835⟩
- Pierre Joanne, Yeranuhi Hovhannisyan, Maximilien Bencze, Marie-Thérèse Daher, Ara Parlakian, et al.. Absence of desmin results in impaired adaptive response to mechanical overloading of skeletal muscle. Frontiers in Cell and Developmental Biology, 2021, 9, pp.662133. ⟨10.3389/fcell.2021.662133⟩. ⟨hal-03343640⟩
- Demetris Koutalianos, Andrie Koutsoulidou, Chrystalla Mytidou, Andrea C Kakouri, Anastasis Oulas, et al.. miR-223-3p and miR-24-3p as novel serum-based biomarkers for myotonic dystrophy type 1. Molecular Therapy - Methods and Clinical Development, 2021, 23, pp.169 - 183. ⟨10.1016/j.omtm.2021.09.007⟩. ⟨hal-03753545⟩
- Sumitava Dastidar, Debanjana Majumdar, Jaitip Tipanee, Kshitiz Singh, Arnaud Klein, et al.. Comprehensive transcriptome-wide analysis of spliceopathy correction of myotonic dystrophy using CRISPR-Cas9 in iPSCs-derived cardiomyocytes. Molecular Therapy, 2021, 29 (11), ⟨10.1016/j.ymthe.2021.08.004⟩. ⟨hal-03410645⟩
- Daniel J Owens, Julien Messéant, Sophie Moog, Mark Viggars, Arnaud Ferry, et al.. Lamin-Related Congenital Muscular Dystrophy Alters Mechanical Signaling and Skeletal Muscle Growth. International Journal of Molecular Sciences, 2020, 22 (1), pp.306. ⟨10.3390/ijms22010306⟩. ⟨hal-03146374⟩
- Nan Zhang, Brittani Bewick, Guangbin Xia, Denis Furling, Tetsuo Ashizawa. A CRISPR-Cas13a Based Strategy That Tracks and Degrades Toxic RNA in Myotonic Dystrophy Type 1. Frontiers in Genetics, 2020, 11, ⟨10.3389/fgene.2020.594576⟩. ⟨hal-03410640⟩
- Stéphanie Bauché, Alain Sureau, Damien Sternberg, John Rendu, Céline Buon, et al.. New recessive mutations in SYT2 causing severe presynaptic congenital myasthenic syndromes. Neurology Genetics, 2020, 6 (6), pp.e534. ⟨10.1212/NXG.0000000000000534⟩. ⟨inserm-03555554⟩
- Linda Heskamp, Kees Okkersen, Marlies van Nimwegen, Marieke Ploegmakers, Guillaume Bassez, et al.. Quantitative Muscle MRI Depicts Increased Muscle Mass after a Behavioral Change in Myotonic Dystrophy Type 1. Radiology, 2020, 297 (1), pp.132-142. ⟨10.1148/radiol.2020192518⟩. ⟨hal-03875206⟩
- Gilles Moulay, Jeanne Lainé, Mégane Lemaître, Masayuki Nakamori, Ichizo Nishino, et al.. Alternative splicing of clathrin heavy chain contributes to the switch from coated pits to plaques. Journal of Cell Biology, 2020, 219 (9), ⟨10.1083/jcb.201912061⟩. ⟨hal-03005118⟩
- Arnaud Ferry, Julien Messéant, Ara Parlakian, Mégane Lemaitre, Pauline Roy, et al.. Desmin prevents muscle wasting, exaggerated weakness and fragility, and fatigue in dystrophic mdx mouse. The Journal of Physiology, 2020, 598 (17), pp.3667-3689. ⟨10.1113/JP279282⟩. ⟨hal-02996889⟩
- A. Stantzou, K. Relizani, S Morales-Gonzalez, C. Gallen, A. Grassin, et al.. Extracellular matrix remodelling is associated with muscle force increase in overloaded mouse plantaris muscle. Neuropathology and Applied Neurobiology, 2020, ⟨10.1111/nan.12655⟩. ⟨hal-02944772⟩
- Derick G Wansink, Geneviève Gourdon, Baziel G M van Engelen, Benedikt Schoser. 248th ENMC International Workshop: Myotonic dystrophies: Molecular approaches for clinical purposes, framing a European molecular research network, Hoofddorp, the Netherlands, 11–13 October 2019. Neuromuscular Disorders, 2020, 30, pp.521 - 531. ⟨10.1016/j.nmd.2020.03.011⟩. ⟨hal-03463004⟩
- Emmanuelle Lagrue, Guillaume Bassez. Author response: A large multicenter study of pediatric myotonic dystrophy type 1 for evidence-based management. Neurology, 2020, 94 (3), pp.146-146. ⟨10.1212/WNL.0000000000008825⟩. ⟨hal-03875201⟩
- Stéphanie Tomé, Geneviève Gourdon. DM1 Phenotype Variability and Triplet Repeat Instability: Challenges in the Development of New Therapies. International Journal of Molecular Sciences, 2020, 21 (2), pp.457. ⟨10.3390/ijms21020457⟩. ⟨hal-03753561⟩
- Laura Muraine, Mona Bensalah, Jamila Dhiab, Gonzalo Cordova, Ludovic Arandel, et al.. Transduction Efficiency of Adeno-Associated Virus Serotypes After Local Injection in Mouse and Human Skeletal Muscle. Human Gene Therapy, In press, ⟨10.1089/hum.2019.173⟩. ⟨hal-02472542⟩
- Teresinha Evangelista, Xavière Lornage, Pierre Carlier, Guillaume Bassez, Guy Brochier, et al.. A Heterozygous Mutation in the Filamin C Gene Causes an Unusual Nemaline Myopathy With Ring Fibers. Journal of Neurology, Neurosurgery and Psychiatry, 2020, 79 (8), pp.908-914. ⟨10.1093/jnen/nlaa052⟩. ⟨hal-03664347⟩
- Ivana Prokic, Belinda S Cowling, Candice Kutchukian, Christine Kretz, Hichem Tasfaout, et al.. Differential physiological role of BIN1 isoforms in skeletal muscle development, function and regeneration. Disease Models & Mechanisms, In press, ⟨10.1242/dmm.044354⟩. ⟨hal-02964521⟩
- Maria Sabater-Arcis, Ariadna Bargiela, Denis Furling, Ruben Artero. miR-7 Restores Phenotypes in Myotonic Dystrophy Muscle Cells by Repressing Hyperactivated Autophagy. Molecular Therapy - Nucleic Acids, 2020, 19, pp.278-292. ⟨10.1016/j.omtn.2019.11.012⟩. ⟨hal-02530984⟩
- Emilie Auxerre-Plantié, Masayuki Nakamori, Yoan Renaud, Aline Huguet, Caroline Choquet, et al.. Straightjacket/α2δ3 deregulation is associated with cardiac conduction defects in myotonic dystrophy type 1. eLife, 2019, 8, pp.e51114. ⟨10.7554/eLife.51114⟩. ⟨hal-02427104⟩
- N. Voermans, R. van der Bilt, J. Ijspeert, J. Hogrel, M. Jeanpierre, et al.. Scapular dyskinesis in myotonic dystrophy type 1: clinical characteristics and genetic investigations. Journal of Neurology, 2019, 266 (12), pp.2987-2996. ⟨10.1007/s00415-019-09494-8⟩. ⟨hal-03875176⟩
- Marie de Antonio, Celine Dogan, Ferroudja Daidj, Bruno Eymard, Jack Puymirat, et al.. The DM-scope registry: a rare disease innovative framework bridging the gap between research and medical care. Orphanet Journal of Rare Diseases, 2019, 14 (1), pp.122. ⟨10.1186/s13023-019-1088-3⟩. ⟨hal-02148794⟩
- Massiré Traoré, Christel Gentil, Chiara Benedetto, Jean-Yves Hogrel, Pierre de La Grange, et al.. An embryonic CaVβ1 isoform promotes muscle mass maintenance via GDF5 signaling in adult mouse. Science Translational Medicine, 2019, 11 (517), ⟨10.1126/scitranslmed.aaw1131⟩. ⟨hal-02382706⟩
- Mei Wang, Wen-Chin Weng, Lauren Stock, Diana Lindquist, Ana Martinez, et al.. Correction of Glycogen Synthase Kinase 3β in Myotonic Dystrophy 1 Reduces the Mutant RNA and Improves Postnatal Survival of DMSXL Mice. Molecular and Cellular Biology, 2019, 39, ⟨10.1128/mcb.00155-19⟩. ⟨hal-03463032⟩
- E. Gargaun, K. Wahbi, R. Ben Yaou, M. Guibaud, G. Solé, et al.. Phenotypic and genomic characterization as predictors of DMD 45 to 55 multi-exon skipping therapy. Neuromuscular Disorders, 2019, 29, pp.S165. ⟨10.1016/j.nmd.2019.06.449⟩. ⟨cea-04414398⟩
- Sarah Cumming, Cecilia Jimenez-Moreno, Kees Okkersen, Stephan Wenninger, Ferroudja Daidj, et al.. Genetic determinants of disease severity in the myotonic dystrophy type 1 OPTIMISTIC cohort. Neurology, 2019, 93 (10), pp.e995-e1009. ⟨10.1212/WNL.0000000000008056⟩. ⟨hal-03875166⟩
- Benedikt Schoser, Federica Montagnese, Guillaume Bassez, Barbara Fossati, Josep Gamez, et al.. Consensus-based care recommendations for adults with myotonic dystrophy type 2. Neurology: Clinical Practice, 2019, 9 (4), pp.343-353. ⟨10.1212/CPJ.0000000000000645⟩. ⟨hal-03875193⟩
- Karim Wahbi, Denis Furling. Cardiovascular manifestations of Myotonic Dystrophy. Trends in Cardiovascular Medicine, 2019, Vol 30 (2020), pp.232-238. ⟨10.1016/j.tcm.2019.06⟩. ⟨hal-02357309⟩
- Ngoc Lu-Nguyen, Arnaud Ferry, Frederick Schnell, Gunnar Hanson, Linda Popplewell, et al.. Functional muscle recovery following dystrophin and myostatin exon splice modulation in aged mdx mice. Human Molecular Genetics, 2019, ⟨10.1093/hmg/ddz125⟩. ⟨hal-03831028⟩
- Mirella Lo Scrudato, Karine Poulard, Célia Sourd, Stéphanie Tomé, Arnaud Klein, et al.. Genome Editing of Expanded CTG Repeats within the Human DMPK Gene Reduces Nuclear RNA Foci in the Muscle of DM1 Mice. Molecular Therapy, 2019, Epub ahead of print. ⟨10.1016/j.ymthe.2019.05.021⟩. ⟨hal-02177548⟩
- Linda Heskamp, Marlies van Nimwegen, Marieke J Ploegmakers, Guillaume Bassez, Jean-Francois Deux, et al.. Lower extremity muscle pathology in myotonic dystrophy type 1 assessed by quantitative MRI. Neurology, 2019, 92, pp.e2803 - e2814. ⟨10.1212/wnl.0000000000007648⟩. ⟨hal-03463112⟩
- Abdallah Fayssoil, Lee S Nguyen, Adam Ogna, Tanya Stojkovic, Paris Meng, et al.. Diaphragm sniff ultrasound: Normal values, relationship with sniff nasal pressure and accuracy for predicting respiratory involvement in patients with neuromuscular disorders. PLoS ONE, 2019, 14 (4), pp.e0214288. ⟨10.1371/journal.pone.0214288⟩. ⟨inserm-02263797⟩
- Yeranuhi Hovhannisyan, Gagik Melikyan, Nathalie Mougenot, Jacqueline Gao-Li, Bertrand Friguet, et al.. Effects of the selective inhibition of proteasome caspase-like activity by CLi a derivative of nor-cerpegin in dystrophic mdx mice. PLoS ONE, 2019, 14 (4), pp.e0215821. ⟨10.1371/journal.pone.0215821⟩. ⟨hal-02124023⟩
- Laura Valentina Renna, Simona Baghai Sain, Christine Voellenkle, Alessandra Perfetti, Matteo Carrara, et al.. Dysregulation of Circular RNAs in Myotonic Dystrophy Type 1. International Journal of Molecular Sciences, 2019, 20 (8), pp.1938. ⟨10.3390/ijms20081938⟩. ⟨hal-02142017⟩
- Emmanuelle Lagrue, Celine Dogan, Marie de Antonio, Frédérique Audic, Nathalie Bach, et al.. A large multicenter study of pediatric myotonic dystrophy type 1 for evidence-based management. Neurology, 2019, 92 (8), pp.e852-e865. ⟨10.1212/WNL.0000000000006948⟩. ⟨hal-02097112⟩
- Guillaume Bassez, Etienne Audureau, Marc Peschanski. Reply: Could weight loss contribute to the improved mobility with metformin in patients with myotonic dystrophy type 1?. Brain - A Journal of Neurology , 2019, 142 (2), pp.e6-e6. ⟨10.1093/brain/awy337⟩. ⟨inserm-04491935⟩
- Sandrine Baghdoyan, Guillaume Bassez, Etienne Audureau, Marc Peschanski. Recherche pharmacologique et cellules souches pluripotentes : du paradigme expérimental novateur à l’essai clinique fructueux. Médecine/Sciences, 2019, 35 (1), pp.26-29. ⟨10.1051/medsci/2018316⟩. ⟨hal-02970141⟩
- Sandra O Braz, Diana M Dinca, Geneviève Gourdon, Mário Gomes-Pereira. Real Time Videomicroscopy and Semiautomated Analysis of Brain Cell Culture Models of Trinucleotide Repeat Expansion Diseases. Methods in Molecular Biology, 2019, Trinucleotide Repeats. Methods and Protocols, 2056, pp.217 - 240. ⟨10.1007/978-1-4939-9784-8_14⟩. ⟨hal-03753559⟩
- Jennifer Morgan, Gillian Butler-Browne, Francesco Muntoni, Ketan Patel, Helge Amthor, et al.. 240th ENMC workshop: The involvement of skeletal muscle stem cells in the pathology of muscular dystrophies 25-27 January 2019, Hoofddorp, The Netherlands. Neuromuscular Disorders, 2019, 29 (9), pp.704-715. ⟨10.1016/j.nmd.2019.07.003⟩. ⟨hal-03201551⟩
- Yves Maury, Pauline Poydenot, Benjamin Brinon, Lea Lesueur, Jacqueline Gide, et al.. Pluripotent Stem Cell-Based Drug Screening Reveals Cardiac Glycosides as Modulators of Myotonic Dystrophy Type 1. iScience, 2019, 11, pp.258-271. ⟨10.1016/j.isci.2018.12.019⟩. ⟨hal-01984689⟩
- Xavière Lornage, Norma B Romero, Claire A. Grosgogeat, Eduardo Malfatti, Sandra Donkervoort, et al.. ACTN2 mutations cause "Multiple structured Core Disease" (MsCD). Acta Neuropathologica, 2019, 137 (3), pp.501-519. ⟨10.1007/s00401-019-01963-8⟩. ⟨hal-03676431⟩
- Montse Olivé, Martin Engvall, Gianina Ravenscroft, Macarena Cabrera-Serrano, Hong Jiao, et al.. Myoglobinopathy is an adult-onset autosomal dominant myopathy with characteristic sarcoplasmic inclusions. Nature Communications, 2019, 10, pp.1396. ⟨10.1038/s41467-019-09111-2⟩. ⟨hal-02110885⟩
- Arnaud F Klein, Miguel A Varela, Ludovic Arandel, Ashling Holland, Naira Naouar, et al.. Peptide-conjugated oligonucleotides evoke long-lasting myotonic dystrophy correction in patient-derived cells and mice. Journal of Clinical Investigation, 2019, 129 (11), pp.4739 - 4744. ⟨10.1172/jci128205⟩. ⟨hal-03753531⟩
- Tetsuo Ashizawa, Cynthia Gagnon, William Groh, Laurie Gutmann, Nicholas Johnson, et al.. Consensus-based care recommendations for adults with myotonic dystrophy type 1. Neurology: Clinical Practice, 2018, 8 (6), pp.507-520. ⟨10.1212/CPJ.0000000000000531⟩. ⟨hal-03875158⟩
- Chantal Sellier, Estefanía Cerro-Herreros, Markus Blatter, Fernande Freyermuth, Angeline Gaucherot, et al.. rbFOX1/MBNL1 competition for CCUG RNA repeats binding contributes to myotonic dystrophy type 1/type 2 differences. Nature Communications, 2018, 9 (1), ⟨10.1038/s41467-018-04370-x⟩. ⟨hal-03339515⟩
- Clement Delacroix, Janek Hyzewicz, Megane Lemaitre, Bertrand Friguet, Zhenlin Li, et al.. Improvement of Dystrophic Muscle Fragility by Short-Term Voluntary Exercise through Activation of Calcineurin Pathway in mdx Mice. American Journal of Pathology, 2018, 188 (11), pp.2662-2673. ⟨10.1016/j.ajpath.2018.07.015⟩. ⟨hal-04443109⟩
- Clement Delacroix, Janek Hyzewicz, Megane Lemaitre, Bertrand Friguet, Zhenlin Li, et al.. Improvement of Dystrophic Muscle Fragility by Short-Term Voluntary Exercise through Activation of Calcineurin Pathway in mdx Mice. American Journal of Pathology, 2018, 188 (11), pp.2662-2673. ⟨10.1016/j.ajpath.2018.07.015⟩. ⟨hal-02134917⟩
- T.N. Brignol, N. Leveziel, C. Dogan, H. Moussu-Haudebourg, G. Bassez. Atteintes rétiniennes dans la DM1 : revue de la littérature et présentation d’un projet basé sur DM-Scope. Journal Français d'Ophtalmologie, 2018, 41 (9), pp.e445-e446. ⟨10.1016/j.jfo.2018.03.016⟩. ⟨hal-03875139⟩
- Guillaume Bassez, Etienne Audureau, Jean-Yves Hogrel, Raphaëlle Arrouasse, Sandrine Baghdoyan, et al.. Improved mobility with metformin in patients with myotonic dystrophy type 1: a randomized controlled trial. Brain - A Journal of Neurology , 2018, 141 (10), pp.2855-2865. ⟨10.1093/brain/awy231⟩. ⟨hal-03875133⟩
- Maximilien Sochala, Raphaël Porcher, Tanya Stojkovic, Henri Marc Bécane, Anthony Behin, et al.. High Risk of Fatal and Nonfatal Venous Thromboembolism in Myotonic Dystrophy. Circulation, 2018, 138 (11), pp.1169-1171. ⟨10.1161/CIRCULATIONAHA.118.035035⟩. ⟨hal-02357318⟩
- Kees Okkersen, Cecilia Jimenez-Moreno, Stephan Wenninger, Ferroudja Daidj, Jeffrey Glennon, et al.. Cognitive behavioural therapy with optional graded exercise therapy in patients with severe fatigue with myotonic dystrophy type 1: a multicentre, single-blind, randomised trial. The Lancet Neurology, 2018, 17 (8), pp.671-680. ⟨10.1016/S1474-4422(18)30203-5⟩. ⟨hal-03875095⟩
- Sandra O Braz, Julien Acquaire, Geneviève Gourdon, Mário Gomes-Pereira. Of Mice and Men: Advances in the Understanding of Neuromuscular Aspects of Myotonic Dystrophy. Frontiers in Neurology, 2018, 9, ⟨10.3389/fneur.2018.00519⟩. ⟨hal-03753310⟩
- Stéphanie Tomé, Elodie Dandelot, Celine Dogan, Alexis Bertrand, David Geneviève, et al.. Unusual association of a unique CAG interruption in 5′ of DM1 CTG repeats with intergenerational contractions and low somatic mosaicism. Human Mutation, 2018, 39 (7), pp.970 - 982. ⟨10.1002/humu.23531⟩. ⟨hal-01870349⟩
- Magdalena Matloka, Arnaud F Klein, Frédérique Rau, Denis Furling. Cells of Matter—In Vitro Models for Myotonic Dystrophy. Frontiers in Neurology, 2018, 9, ⟨10.3389/fneur.2018.00361⟩. ⟨hal-03830776⟩
- Karim Wahbi, Raphaël Porcher, Pascal Laforêt, Abdallah Fayssoil, Henri Marc Bécane, et al.. Development and Validation of a New Scoring System to Predict Survival in Patients With Myotonic Dystrophy Type 1. JAMA neurology, 2018, 75 (5), pp.573. ⟨10.1001/jamaneurol.2017.4778⟩. ⟨hal-03830888⟩
- Feriel Azibani, Astrid Brull, Ludovic Arandel, Maud Beuvin, Isabelle Nelson, et al.. Gene Therapy via Trans-Splicing for LMNA-Related Congenital Muscular Dystrophy. Molecular Therapy - Nucleic Acids, 2018, 10, pp.376 - 386. ⟨10.1016/j.omtn.2017.12.012⟩. ⟨hal-03269960⟩
- Judith van Vliet, Alide Tieleman, Baziel G.M. van Engelen, Guillaume Bassez, Laurent Servais, et al.. Hearing impairment in patients with myotonic dystrophy type 2. Neurology, 2018, 90 (7), pp.e615-e622. ⟨10.1212/WNL.0000000000004963⟩. ⟨hal-03875067⟩
- Simone Birnbaum, Jean-Yves Hogrel, Raphael Porcher, Pierre Portero, Bernard Clair, et al.. The benefits and tolerance of exercise in myasthenia gravis (MGEX): study protocol for a randomised controlled trial. Trials, 2018, 19, pp.49. ⟨10.1186/s13063-017-2433-2⟩. ⟨hal-01708023⟩
- Sumitava Dastidar, Simon Ardui, Kshitiz Singh, Debanjana Majumdar, Nisha Nair, et al.. Efficient CRISPR/Cas9-mediated editing of trinucleotide repeat expansion in myotonic dystrophy patient-derived iPS and myogenic cells. Nucleic Acids Research, 2018, 46, pp.8275 - 8298. ⟨10.1093/nar/gky548⟩. ⟨hal-02357315⟩
- Delphine Trochet, Bernard Prudhon, Maud Beuvin, Cécile Peccate, Stéphanie Lorain, et al.. Allele‐specific silencing therapy for Dynamin 2‐related dominant centronuclear myopathy. EMBO Molecular Medicine, 2018, 10 (2), pp.239-253. ⟨10.15252/emmm.201707988⟩. ⟨hal-02000303⟩
- Robin Deloux, Cynthia Tannous, Arnaud Ferry, Zhenlin Li, Mathias Mericskay. Aged Nicotinamide Riboside Kinase 2 Deficient Mice Present an Altered Response to Endurance Exercise Training. Frontiers in Physiology, 2018, 9, pp.1290. ⟨10.3389/fphys.2018.01290⟩. ⟨hal-01884625⟩
- Milena Rizzo, Pascale Beffy, Renata Del Carratore, Alessandra Falleni, Virginia Pretini, et al.. Activation of the interferon type I response rather than autophagy contributes to myogenesis inhibition in congenital DM1 myoblasts. Cell Death and Disease, 2018, 9, ⟨10.1038/s41419-018-1080-1⟩. ⟨hal-02357317⟩
- Elodie Dandelot, Geneviève Gourdon. The flash-small-pool PCR: how to transform blotting and numerous hybridization steps into a simple denatured PCR. Biotechniques, 2018, 64 (6), pp.262 - 265. ⟨10.2144/btn-2018-0035⟩. ⟨hal-03753523⟩
- Delphine Trochet, Bernard Prudhon, Maud Beuvin, Cécile Peccate, Stéphanie Lorain, et al.. Allele-specific silencing therapy for Dynamin 2-related dominant centronuclear myopathy. Circulation. Arrhythmia and electrophysiology, 2017, 10 (12), pp.428-431. ⟨10.15252/emmm.201707988⟩. ⟨hal-04001377⟩
- Maria Grazia Biferi, Mathilde Cohen-Tannoudji, Ambra Cappelletto, Benoit Giroux, Marianne Roda, et al.. A New AAV10-U7-Mediated Gene Therapy Prolongs Survival and Restores Function in an ALS Mouse Model. Molecular Therapy, 2017, 25 (9), pp.2038-2052. ⟨10.1016/j.ymthe.2017.05.017⟩. ⟨hal-03829068⟩
- Géraldine Sicot, Laurent Servais, Diana Dinca, Axelle M Leroy, Cynthia Prigogine, et al.. Downregulation of the Glial GLT1 Glutamate Transporter and Purkinje Cell Dysfunction in a Mouse Model of Myotonic Dystrophy. Cell Reports, 2017, 19 (13), pp.2718-2729. ⟨10.1016/j.celrep.2017.06.006⟩. ⟨hal-03164812⟩
- Caroline Chong-Nguyen, Karim Wahbi, Vincent Algalarrondo, Henri Marc Bécane, Hélène Radvanyi-Hoffman, et al.. Association Between Mutation Size and Cardiac Involvement in Myotonic Dystrophy Type 1. Circulation: Cardiovascular Genetics, 2017, 10 (3), ⟨10.1161/CIRCGENETICS.116.001526⟩. ⟨hal-03830988⟩
- Mário Gomes-Pereira, Darren G Monckton. Ethidium Bromide Modifies The Agarose Electrophoretic Mobility of CAG•CTG Alternative DNA Structures Generated by PCR. Frontiers in Cellular Neuroscience, 2017, 11, ⟨10.3389/fncel.2017.00153⟩. ⟨hal-03164849⟩
- Floriane Lacour, Elsa Vezin, C. Florian Bentzinger, Marie-Claude Sincennes, Lorenzo Giordani, et al.. R-spondin1 Controls Muscle Cell Fusion through Dual Regulation of Antagonistic Wnt Signaling Pathways. Cell Reports, 2017, 18 (10), pp.2320 - 2330. ⟨10.1016/j.celrep.2017.02.036⟩. ⟨hal-04765990⟩
- Simon Guiraud, Tiffany Migeon, Arnaud Ferry, Zhiyong Chen, Souhila Ouchelouche, et al.. HANAC Col4a1 Mutation in Mice Leads to Skeletal Muscle Alterations due to a Primary Vascular Defect. American Journal of Pathology, 2017, 187 (3), pp.505-516. ⟨10.1016/j.ajpath.2016.10.020⟩. ⟨hal-03831011⟩
- Floriane Lacour, Elsa Vezin, C. Florian Bentzinger, Marie-Claude Sincennes, Lorenzo Giordani, et al.. R-spondin1 Controls Muscle Cell Fusion through Dual Regulation of Antagonistic Wnt Signaling Pathways. Cell Reports, 2017, 18 (10), pp.2320-2330. ⟨10.1016/j.celrep.2017.02.036⟩. ⟨hal-02486025⟩
- Suzanne Rzuczek, Lesley Colgan, Yoshio Nakai, Michael Cameron, Denis Furling, et al.. Precise small-molecule recognition of a toxic CUG RNA repeat expansion. Nature Chemical Biology, 2017, 13 (2), pp.188-193. ⟨10.1038/nchembio.2251⟩. ⟨hal-03830973⟩
- Marielle Brockhoff, Nathalie Rion, Kathrin Chojnowska, Tatiana Wiktorowicz, Christopher Eickhorst, et al.. Targeting deregulated AMPK/mTORC1 pathways improves muscle function in myotonic dystrophy type I. Journal of Clinical Investigation, 2017, 127 (2), pp.549-563. ⟨10.1172/JCI89616⟩. ⟨hal-03830899⟩
- Ellen L. van Agtmaal, Laurène M. André, Marieke Willemse, Sarah A. Cumming, Ingeborg D.G. van Kessel, et al.. CRISPR/Cas9-Induced (CTG⋅CAG)n Repeat Instability in the Myotonic Dystrophy Type 1 Locus: Implications for Therapeutic Genome Editing. Molecular Therapy, 2017, 25 (1), pp.24 - 43. ⟨10.1016/j.ymthe.2016.10.014⟩. ⟨hal-01429815⟩
- Vanessa Ueberschlag-Pitiot, Amalia Stantzou, Julien Messéant, Megane Lemaitre, Daniel Owens, et al.. Gonad-related factors promote muscle performance gain during postnatal development in male and female mice. AJP - Endocrinology and Metabolism, 2017, 313 (1), pp.E12-E25. ⟨10.1152/ajpendo.00446.2016⟩. ⟨hal-03677800⟩
- Genevieve Gourdon, Giovanni Meola. Myotonic Dystrophies: State of the Art of New Therapeutic Developments for the CNS. Frontiers in Cellular Neuroscience, 2017, 11, pp.101. ⟨10.3389/fncel.2017.00101⟩. ⟨hal-03832511⟩
- A. Malerba, P. Klein, H. Bachtarzi, S A Jarmin, G. Cordova, et al.. PABPN1 gene therapy for oculopharyngeal muscular dystrophy. Nature Communications, 2017, 8, pp.14848. ⟨10.1038/ncomms14848⟩. ⟨hal-01959996⟩
- Robin Deloux, Damien Vitiello, Nathalie Mougenot, Philippe Noirez, Zhenlin Li, et al.. Voluntary Exercise Improves Cardiac Function and Prevents Cardiac Remodeling in a Mouse Model of Dilated Cardiomyopathy. Frontiers in Physiology, 2017, 8, pp.899. ⟨10.3389/fphys.2017.00899⟩. ⟨hal-02650844⟩
- Ludovic Arandel, Micaela Polay Espinoza, Magdalena Matloka, Audrey Bazinet, Damily de Dea Diniz, et al.. Immortalized human myotonic dystrophy muscle cell lines to assess therapeutic compounds. Disease Models & Mechanisms, 2017, 10 (4), pp.487-497. ⟨10.1242/dmm.027367⟩. ⟨hal-01519721⟩
- Ivana Dinulovic, Regula Furrer, Markus Beer, Arnaud Ferry, Bettina Cardel, et al.. Muscle PGC-1α modulates satellite cell number and proliferation by remodeling the stem cell niche. Skeletal Muscle, 2016, 6, pp.39. ⟨10.1186/s13395-016-0111-9⟩. ⟨hal-01449364⟩
- Pauline Roy, Fredérique Rau, Julien Ochala, Julien Messéant, Bodvael Fraysse, et al.. Dystrophin restoration therapy improves both the reduced excitability and the force drop induced by lengthening contractions in dystrophic mdx skeletal muscle. Skeletal Muscle, 2016, 6 (1), pp.23. ⟨10.1186/s13395-016-0096-4⟩. ⟨hal-01357455⟩
- Fernande Freyermuth, Frédérique Rau, Yosuke Kokunai, Thomas Linke, Chantal Sellier, et al.. Splicing misregulation of SCN5A contributes to cardiac-conduction delay and heart arrhythmia in myotonic dystrophy. Nature Communications, 2016, 7, pp.11067. ⟨10.1038/ncomms11067⟩. ⟨hal-01301863⟩
- Sandro Alves, Thibaut Marais, Maria-Grazia Biferi, Denis Furling, Martina Marinello, et al.. Lentiviral vector-mediated overexpression of mutant ataxin-7 recapitulates SCA7 pathology and promotes accumulation of the FUS/TLS and MBNL1 RNA-binding proteins. Molecular Neurodegeneration, 2016, 11, pp.58. ⟨10.1186/s13024-016-0123-2⟩. ⟨hal-01371536⟩
- Marie-Elodie Cattin, Arnaud Ferry, Alban Vignaud, Nathalie Mougenot, Adeline Jacquet, et al.. Mutation in lamin A/C sensitizes the myocardium to exercise-induced mechanical stress but has no effect on skeletal muscles in mouse. Neuromuscular Disorders, 2016, ⟨10.1016/j.nmd.2016.05.010⟩. ⟨hal-01329664⟩
- Amalia Stantzou, Vanessa Ueberschlag-Pitiot, Rémi Thomasson, Denis Furling, Anne Bonnieu, et al.. The effect of constitutive inactivation of the myostatin gene on the gain in muscle strength during postnatal growth in two murine models. Muscle & Nerve, 2016, 55 (2), pp.254-261. ⟨10.1002/mus.25220⟩. ⟨hal-01346093⟩
- Arnaud Ferry, Rachid Benchaouir, Pierre Joanne, Rachel A. Peat, Nathalie Mougenot, et al.. Effect of voluntary physical activity initiated at age 7 months on skeletal hindlimb and cardiac muscle function in mdx mice of both genders. Muscle & Nerve, 2015, 52 (5), pp.788-794. ⟨10.1002/mus.24604⟩. ⟨hal-01545442⟩
- Jordan Blondelle, Yusuke Ohno, Vincent Gache, Stéphane Guyot, Sébastien Storck, et al.. HACD1 , a regulator of membrane composition and fluidity, promotes myoblast fusion and skeletal muscle growth. Journal of molecular cell biology, 2015, 7 (5), pp.429-440. ⟨10.1093/jmcb/mjv049⟩. ⟨hal-02290631⟩
- C. Moinard, S. Le Plenier, P. Noirez, Béatrice Morio, D. Bonnefont-Rousselot, et al.. Citrulline Supplementation Induces Changes in Body Composition and Limits Age-Related Metabolic Changes in Healthy Male Rats. Journal of Nutrition, 2015, 145 (7), pp.1429-37. ⟨10.3945/jn.114.200626⟩. ⟨hal-01850474⟩
- Arnaud Ferry, Ara Parlakian, Pierre Joanne, Bodvael Fraysse, Takouhie Mgrditchian, et al.. Mechanical Overloading Increases Maximal Force and Reduces Fragility in Hind Limb Skeletal Muscle from Mdx Mouse. American Journal of Pathology, 2015, 185 (7), pp.2012-2024. ⟨10.1016/j.ajpath.2015.03.027⟩. ⟨hal-01545443⟩
- Carole Henique, Abdelhak Mansouri, Eliska Vavrova, Veronique Lenoir, Arnaud Ferry, et al.. Increasing mitochondrial muscle fatty acid oxidation induces skeletal muscle remodeling toward an oxidative phenotype. FASEB Journal, 2015, 29 (6), pp.2473-2483. ⟨10.1096/fj.14-257717⟩. ⟨hal-02122695⟩
- Mirella Lo Scrudato, Samia Martin, Geneviève Gourdon, Denis Furling, Ana Buj-Bello. 576. A Novel Gene Editing-Based Strategy for Myotonic Dystrophy Type 1. Molecular Therapy, 2015, 23, pp.S229. ⟨10.1016/S1525-0016(16)34185-5⟩. ⟨hal-03159325⟩
- Aurélie Goyenvalle, Graziella Griffith, Arran Babbs, Samir El Andaloussi, Kariem Ezzat, et al.. Functional correction in mouse models of muscular dystrophy using exon-skipping tricyclo-DNA oligomers. Nature Medicine, 2015, 21 (3), pp.270-275. ⟨10.1038/nm.3765⟩. ⟨hal-02407968⟩
- Laetitia Ramanoudjame, Claire Rocancourt, Jeanne Lainé, Arnaud Klein, Lucette Joassard, et al.. Two novel COLVI long chains in zebrafish that are essential for muscle development. Human Molecular Genetics, 2015, 24 (23), pp.6624-6639. ⟨10.1093/hmg/ddv368⟩. ⟨hal-01594462⟩
- Frédérique Rau, Jeanne Lainé, Laetitita Ramanoudjame, Arnaud Ferry, Ludovic Arandel, et al.. Abnormal splicing switch of DMD's penultimate exon compromises muscle fibre maintenance in myotonic dystrophy. Nature Communications, 2015, 6 (1), pp.7205. ⟨10.1038/ncomms8205⟩. ⟨hal-01162385⟩
- Arnaud Ferry, Melanie Schuh, Ara Parlakian, Takouhie Mgrditchian, Nicolas Valnaud, et al.. Myofiber Androgen Receptor Promotes Maximal Mechanical Overload-Induced Muscle Hypertrophy and Fiber Type Transition in Male Mice. Endocrinology, 2014, 155 (12), pp.4739-4748. ⟨10.1210/en.2014-1195⟩. ⟨hal-01545451⟩
- Arnaud Ferry, Pierre Joanne, Wahiba Hadj-Said, Alban Vignaud, Alain Lilienbaum, et al.. Advances in the understanding of skeletal muscle weakness in murine models of diseases affecting nerve-evoked muscle activity, motor neurons, synapses and myofibers. Neuromuscular Disorders, 2014, 24 (11), pp.960-972. ⟨10.1016/j.nmd.2014.06.001⟩. ⟨hal-01545452⟩
- Zhenlin Li, Ara Parlakian, Dario Coletti, Sonia Alonso-Martin, Christophe Hourde, et al.. Synemin acts as a regulator of signalling molecules during skeletal muscle hypertrophy. Journal of Cell Science, 2014, 127 (21), pp.4589-4601. ⟨10.1242/jcs.143164⟩. ⟨hal-01545450⟩
- Sandrine Loron, Arnaud Dulac, Olivier Jegaden, Tristan Ferry. Large vegetation in a 60-year-old man with Enterococcus faecalis cardiac implantable electronic device infection. BMJ Case Reports, 2014, 2014, ⟨10.1136/bcr-2014-206907⟩. ⟨hal-01911488⟩
- Roscoe Klinck, Angélique Fourrier, Philippe Thibault, Johanne Toutant, Mathieu Durand, et al.. RBFOX1 Cooperates with MBNL1 to Control Splicing in Muscle, Including Events Altered in Myotonic Dystrophy Type 1. PLoS ONE, 2014, 9 (9), pp.e107324. ⟨10.1371/journal.pone.0107324⟩. ⟨hal-01365880⟩
- Bodvaël Fraysse, Alban Vignaud, Bourama Fane, Mélanie Schuh, Gillian S. Butler-Browne, et al.. Acute effect of androgens on maximal force-generating capacity and electrically evoked calcium transient in mouse skeletal muscles. Steroids, 2014, 87, pp.6-11. ⟨10.1016/j.steroids.2014.05.005⟩. ⟨hal-02881144⟩
- Etienne Mouisel, Karima Relizani, Laurence Mille-Hamard, Raphael Denis, Christophe Hourde, et al.. Myostatin is a key mediator between energy metabolism and endurance capacity of skeletal muscle. AJP - Regulatory, Integrative and Comparative Physiology, 2014, 307 (4), pp.R444-R454. ⟨10.1152/ajpregu.00377.2013⟩. ⟨hal-01545449⟩
- Louise Lantier, Joachim Fentz, Rémi Mounier, Jocelyne Leclerc, Jonas T. Treebak, et al.. AMPK controls exercise endurance, mitochondrial oxidative capacity, and skeletal muscle integrity.. FASEB Journal, 2014, 28 (7), pp.3211-24. ⟨10.1096/fj.14-250449⟩. ⟨inserm-00979373⟩
- Mário Gomes-Pereira, J. Hilley, F. Morales, B. Adam, H. James, et al.. Disease-associated CAG{middle dot}CTG triplet repeats expand rapidly in non-dividing mouse cells, but cell cycle arrest is insufficient to drive expansion. Nucleic Acids Research, 2014, 42 (11), pp.7047-7056. ⟨10.1093/nar/gku285⟩. ⟨hal-03164860⟩
- Stéphane Vassilopoulos, Christel Gentil, Jeanne Lainé, Pierre-Olivier Buclez, Agathe Franck, et al.. Actin scaffolding by clathrin heavy chain is required for skeletal muscle sarcomere organization. Journal of Cell Biology, 2014, 205 (3), pp.377-393. ⟨10.1083/jcb.201309096⟩. ⟨hal-02453865⟩
- Iori Sakakibara, Marc Santolini, Arnaud Ferry, Vincent Hakim, Pascal Maire. Six Homeoproteins and a Iinc-RNA at the Fast MYH Locus Lock Fast Myofiber Terminal Phenotype. PLoS Genetics, 2014, 10 (5), pp.e1004386. ⟨10.1371/journal.pgen.1004386⟩. ⟨hal-01342678⟩
- Florian A Britto, Gwenaelle Begue, Bernadette Rossano, Aurélie Docquier, Barbara Vernus, et al.. REDD1 deletion prevents dexamethasone-induced skeletal muscle atrophy. AJP - Endocrinology and Metabolism, 2014, 307 (11), pp.E983-E993. ⟨10.1152/ajpendo.00234.2014⟩. ⟨hal-01635920⟩
- Stéphanie Tomé, Annie Nicole, Mario Gomes-Pereira, Genevieve Gourdon. Non-Radioactive Detection of Trinucleotide Repeat Size Variability. PLoS Currents, 2014, ⟨10.1371/currents.md.ad50113b899fa1352ce70c087eead706⟩. ⟨hal-03545527⟩
- Florent Valour, Agathe Sénéchal, Céline Dupieux, Judith Karsenty, Sébastien Lustig, et al.. Actinomycosis: etiology, clinical features, diagnosis, treatment, and management. Infection and Drug Resistance, 2014, 7, pp.183-197. ⟨10.2147/IDR.S39601⟩. ⟨hal-01908879⟩
- Belinda S Cowling, Thierry Chevremont, Ivana Prokic, Christine Kretz, Arnaud Ferry, et al.. Reducing dynamin 2 expression rescues X-linked centronuclear myopathy. Journal of Clinical Investigation, 2014, 124 (3), pp.1350-1363. ⟨10.1172/JCI71206⟩. ⟨hal-01329329⟩
- Laurent Saint-André, Julien Sainte-Marie, Sophie Leguédois, Bruno Ferry, Francois Lafolie, et al.. Les avancées de la recherche dans le domaine de la modélisation des interactions sol-arbre. Revue forestière française, 2014, 66 (4), pp.479-490. ⟨10.4267/2042/56561⟩. ⟨hal-01225257⟩
- K. Relizani, E. Mouisel, B. Giannesini, C. Hourde, K. Patel, et al.. Blockade of ActRIIB signaling triggers muscle fatigability and metabolic myopathy. Molecular Therapy, 2014, 22 (8), pp.1423-1433. ⟨10.1038/mt.2014.90⟩. ⟨hal-02881194⟩
- Pierre Deharo, Mathieu Pankert, Guillaume Bonnet, Jacques Quilici, Clemence Bassez, et al.. Body mass index has no impact on platelet inhibition induced by ticagrelor after acute coronary syndrome, conversely to prasugrel. International Journal of Cardiology, 2014, 176 (3), pp.1200-1202. ⟨10.1016/j.ijcard.2014.07.228⟩. ⟨hal-02633519⟩
- Morgane Gauthier, Antoine Marteyn, Jérôme Alexandre Denis, Michel Cailleret, Karine Giraud-Triboult, et al.. A defective Krab-domain zinc-finger transcription factor contributes to altered myogenesis in myotonic dystrophy type 1.. Human Molecular Genetics, 2013, 22 (25), pp.5188-98. ⟨10.1093/hmg/ddt373⟩. ⟨inserm-00952767⟩
- X. Deffieux, L. Sentilhes, D. Savary, V. Letouzey, M. Marcelli, et al.. Indications de la cure du prolapsus génital par voie vaginale avec prothèse : consensus d’experts du Collège national des gynécologues et obstétriciens français (CNGOF). Journal de Gynécologie Obstétrique et Biologie de la Reproduction, 2013, 42 (7), pp.628 - 638. ⟨10.1016/j.jgyn.2013.08.018⟩. ⟨hal-01782583⟩
- Rémi Mounier, Marine Théret, Ludovic Arnold, Sylvain Cuvellier, Laurent Bultot, et al.. AMPKα1 Regulates Macrophage Skewing at the Time of Resolution of Inflammation during Skeletal Muscle Regeneration.. Cell Metab, 2013, 18 (2), pp.251-64. ⟨10.1016/j.cmet.2013.06.017⟩. ⟨inserm-00857933v2⟩
- Christophe Hourde, Pierre Joanne, Philippe Noirez, Onnik Agbulut, Gillian Butler-Browne, et al.. Protective effect of female gender-related factors on muscle force-generating capacity and fragility in the dystrophic mdx mouse. Muscle & Nerve, 2013, 48 (1), pp.68-75. ⟨10.1002/mus.23700⟩. ⟨hal-01545455⟩
- P. Lucot, X. Fritel, P. Debodinance, G. Bader, Michel Cosson, et al.. Étude randomisée comparant la promontofixation cœlioscopique à la chirurgie prothétique par voie vaginale pour le traitement des cystocèles : PROSPERE (PROSthetic PElvic organ prolapse REpair). Journal de Gynécologie Obstétrique et Biologie de la Reproduction, 2013, 42 (4), pp.334 - 341. ⟨10.1016/j.jgyn.2013.03.012⟩. ⟨hal-01782144⟩
- Christophe Hourde, Pierre Joanne, Fadia Medja, Nathalie Mougenot, Adeline Jacquet, et al.. Voluntary Physical Activity Protects from Susceptibility to Skeletal Muscle Contraction-Induced Injury But Worsens Heart Function in mdx Mice. American Journal of Pathology, 2013, 182 (5), pp.1509-1518. ⟨10.1016/j.ajpath.2013.01.020⟩. ⟨hal-01545454⟩
- Leonela Amoasii, Karim Hnia, Gaëtan Chicanne, Andreas Brech, Belinda S Cowling, et al.. Myotubularin and PtdIns3P remodel the sarcoplasmic reticulum in muscle in vivo.. Journal of Cell Science, 2013, 126 (Pt 8), pp.1806-19. ⟨10.1242/jcs.118505⟩. ⟨inserm-01012056⟩
- Pierre Joanne, Oussama Chourbagi, Christophe Hourdé, Arnaud Ferry, Gillian Butler-Browne, et al.. Viral-mediated expression of desmin mutants to create mouse models of myofibrillar myopathy.. Skeletal Muscle, 2013, 3 (1), pp.4. ⟨10.1186/2044-5040-3-4⟩. ⟨inserm-00800537⟩
- E. Schirwis, O. Agbulut, Nathalie Vadrot, E. Mouisel, C. Hourde, et al.. The beneficial effect of myostatin deficiency on maximal muscle force and power is attenuated with age. Experimental Gerontology, 2013, 48 (2), pp.183-190. ⟨10.1016/j.exger.2012.11.008⟩. ⟨hal-01545453⟩
- Pierre Deharo, Clemence Bassez, Guillaume Bonnet, Mathieu Pankert, Jacques Quilici, et al.. Prasugrel versus ticagrelor in acute coronary syndrome: A randomized comparison. International Journal of Cardiology, 2013, 170 (2), pp.E21-E22. ⟨10.1016/j.ijcard.2013.10.043⟩. ⟨hal-02652055⟩
- Arnaud Lejeune, Hakim Boudaoud, Michel Potier-Ferry, Isabelle Charpentier, Hamid Zahrouni. Automatic solver for non-linear partial differential equations with implicit local laws: Application to unilateral contact. International Journal for Numerical Methods in Engineering, 2013, 94, pp.850-867. ⟨10.1002/nme.4483⟩. ⟨hal-00982813⟩
- Denis Vallese, Elisa Negroni, Stéphanie Duguez, Arnaud Ferry, Capucine Trollet, et al.. The Rag2⁻Il2rb⁻Dmd⁻ mouse: a novel dystrophic and immunodeficient model to assess innovating therapeutic strategies for muscular dystrophies.. Molecular Therapy, 2013, 21 (10), pp.1950-7. ⟨10.1038/mt.2013.186⟩. ⟨pasteur-01489680⟩
- Pierre Joanne, Christophe Hourde, Julien Ochala, Yvain Cauderan, Fadia Medja, et al.. Impaired Adaptive Response to Mechanical Overloading in Dystrophic Skeletal Muscle. PLoS ONE, 2012, 7 (4), pp.e35346. ⟨10.1371/journal.pone.0035346⟩. ⟨hal-01545458⟩
- Wahiba Hadj-Said, Marie Bangratz, Alban Vignaud, Arnaud Chatonnet, Gillian Butler-Browne, et al.. Effect of locomotor training on muscle performance in the context of nerve-muscle communication dysfunction. Muscle & Nerve, 2012, 45 (4), pp.567-577. ⟨10.1002/mus.22332⟩. ⟨hal-01545457⟩
- François-Xavier Laurent, Alain Sureau, Arnaud F Klein, François Trouslard, Erwan Gasnier, et al.. New function for the RNA helicase p68/DDX5 as a modifier of MBNL1 activity on expanded CUG repeats. Nucleic Acids Research, 2012, 40 (7), pp.3159-3171. ⟨10.1093/nar/gkr1228⟩. ⟨hal-03001690⟩
- Arnaud Lejeune, Fabien Béchet, Hakim Boudaoud, Norman Mathieu, Michel Potier-Ferry. Object-oriented design to automate a high order non-linear solver based on asymptotic numerical method. Advances in Engineering Software, 2012, 48, pp.70-88. ⟨10.1016/j.advengsoft.2012.02.012⟩. ⟨hal-03223636⟩
- Leonela Amoasii, Dimitri L. Bertazzi, Hélène Tronchère, Karim Hnia, Gaëtan Chicanne, et al.. Phosphatase-dead myotubularin ameliorates X-linked centronuclear myopathy phenotypes in mice.. PLoS Genetics, 2012, 8 (10), pp.e1002965. ⟨10.1371/journal.pgen.1002965⟩. ⟨inserm-01011824⟩
- Mathieu Rederstorff, Perrine Castets, Sandrine Arbogast, Jeanne Lainé, Stéphane Vassilopoulos, et al.. Increased Muscle Stress-Sensitivity Induced by Selenoprotein N Inactivation in Mouse: A Mammalian Model for SEPN1-Related Myopathy. PLoS ONE, 2011, 6 (8), ⟨10.1371/journal.pone.0023094⟩. ⟨hal-01716017⟩
- Frédérique Rau, Fernande Freyermuth, Charlotte Fugier, Jean-Philippe Villemin, Marie-Christine Fischer, et al.. Misregulation of miR-1 processing is associated with heart defects in myotonic dystrophy. Nature Structural and Molecular Biology, 2011, 18 (7), pp.840-845. ⟨10.1038/nsmb.2067⟩. ⟨hal-03339571⟩
- Belinda S Cowling, Anne Toussaint, Leonela Amoasii, Pascale Koebel, Arnaud Ferry, et al.. Increased Expression of Wild-Type or a Centronuclear Myopathy Mutant of Dynamin 2 in Skeletal Muscle of Adult Mice Leads to Structural Defects and Muscle Weakness. American Journal of Pathology, 2011, 178 (5), pp.2224-2235. ⟨10.1016/j.ajpath.2011.01.054⟩. ⟨hal-04974247⟩
- Helene Tran, Nathalie Gourrier, Camille Lemercier-Neuillet, Claire-Marie Dhaenens, Audrey Vautrin, et al.. Analysis of Exonic Regions Involved in Nuclear Localization, Splicing Activity, and Dimerization of Muscleblind-like-1 Isoforms. Journal of Biological Chemistry, 2011, 286 (18), pp.16435 - 16446. ⟨10.1074/jbc.M110.194928⟩. ⟨hal-01738403⟩
- Perrine Castets, Anne T. Bertrand, Maud Beuvin, Arnaud Ferry, Fabien Le Grand, et al.. Satellite cell loss and impaired muscle regeneration in selenoprotein N deficiency.. Human Molecular Genetics, 2011, 20 (4), pp.694-704. ⟨10.1093/hmg/ddq515⟩. ⟨hal-00561402⟩
- Andrie Koutsoulidou, Nikolaos Mastroyiannopoulos, Denis Furling, James Uney, Leonidas Phylactou. Expression of miR-1, miR-133a, miR-133b and miR-206 increases during development of human skeletal muscle.. BMC Developmental Biology, 2011, 11 (1), pp.34. ⟨10.1186/1471-213X-11-34⟩. ⟨inserm-00668415⟩
- Florent Hubé, Guillaume Velasco, Jérôme Rollin, Denis Furling, Claire Francastel. Steroid receptor RNA activator protein binds to and counteracts SRA RNA-mediated activation of MyoD and muscle differentiation. Nucleic Acids Research, 2011, 39 (2), pp.513-525. ⟨10.1093/nar/gkq833⟩. ⟨hal-02127321⟩
- Arnaud Serry. Dynamiques du transport maritime en Baltique orientale. Territoire en mouvement. Revue de Géographie et d'Aménagement, 2011, 10, pp.36-48. ⟨10.4000/tem.1089⟩. ⟨hal-01724153⟩
- Virginie François, Arnaud F Klein, Cyriaque Beley, Arnaud Jollet, Camille Lemercier, et al.. Selective silencing of mutated mRNAs in DM1 by using modified hU7-snRNAs. Nature Structural and Molecular Biology, 2011, 18 (1), pp.85-87. ⟨10.1038/nsmb.1958⟩. ⟨hal-03002664⟩
- M. Ageron, J.A. Aguilar, Imen Al Samarai, A. Albert, F. Ameli, et al.. ANTARES: the first undersea neutrino telescope. Nuclear Instruments and Methods in Physics Research Section A: Accelerators, Spectrometers, Detectors and Associated Equipment, 2011, 656, pp.11-38. ⟨10.1016/j.nima.2011.06.103⟩. ⟨in2p3-00593848⟩
- Anne-Cécile Durieux, Alban Vignaud, Bernard Prudhon, Mai Thao Viou, Maud Beuvin, et al.. A centronuclear myopathy-dynamin 2 mutation impairs skeletal muscle structure and function in mice. Human Molecular Genetics, 2010, 19 (24), pp.4820-4836. ⟨10.1093/hmg/ddq413⟩. ⟨hal-02451060⟩
- P. Castets, A.T. Bertrand, M. Beuvin, A. Ferry, F. Le Grand, et al.. O.3 Satellite cell loss is the pathomechanism leading to muscle atrophy in selenoprotein N deficiency. Neuromuscular Disorders, 2010, 20 (9-10), pp.598. ⟨10.1016/J.NMD.2010.07.012⟩. ⟨hal-02935581⟩
- Chantal Sellier, Frédérique Rau, Yilei Liu, Flora Tassone, Renate Hukema, et al.. Sam68 sequestration and partial loss of function are associated with splicing alterations in FXTAS patients. EMBO Journal, 2010, 29 (7), pp.1248-1261. ⟨10.1038/emboj.2010.21⟩. ⟨hal-03339578⟩
- Ara Parlakian, Iman Gomaa, Sounkary Solly, Ludovic Arandel, Alka Mahale, et al.. Skeletal Muscle Phenotypically Converts and Selectively Inhibits Metastatic Cells in Mice. PLoS ONE, 2010, 5 (2), pp.e9299. ⟨10.1371/journal.pone.0009299⟩. ⟨hal-03832536⟩
- France Piétri-Rouxel, Christel Gentil, Stéphane Vassilopoulos, Dominique Baas, Etienne Mouisel, et al.. DHPR alpha1S subunit controls skeletal muscle mass and morphogenesis.. EMBO Journal, 2010, 29 (3), pp.643-54. ⟨10.1038/emboj.2009.366⟩. ⟨inserm-00515849⟩
- Brigitte Blondet, Gilles Carpentier, Arnaud Ferry, Arnaud Chatonnet, José Courty. Localization of butyrylcholinesteraseat the neuromuscular junction of normal and acetylcholinesterase knockout mice. Journal of Histochemistry and Cytochemistry, 2010, 58 (12), pp.1075-1082. ⟨10.1369/jhc.2010.956623⟩. ⟨hal-02665164⟩
- Fabien Béchet, Arnaud Lejeune, Michel Ferry. Taylor series to solve friction problems. Comptes Rendus. Mécanique, 2010, 338 (6), pp.327 - 332. ⟨hal-02300155⟩
- France Piétri-Rouxel, Christel Gentil, Stéphane Vassilopoulos, Dominique Baas, Etienne Mouisel, et al.. DHPR α1S subunit controls skeletal muscle mass and morphogenesis. EMBO Journal, 2009, 29, pp.643 - 654. ⟨ensl-00817432⟩
- Valérie Risson, Laetitia Mazelin, Mila Roceri, Hervé Sanchez, Vincent Moncollin, et al.. Muscle inactivation of mTOR causes metabolic and dystrophin defects leading to severe myopathy. Journal of Cell Biology, 2009, 187 (6), pp.859-874. ⟨10.1083/jcb.200903131⟩. ⟨hal-02126916⟩
- S. Mulders, W. van den Broek, T. Wheeler, H. Croes, P. van Kuik-Romeijn, et al.. Triplet-repeat oligonucleotide-mediated reversal of RNA toxicity in myotonic dystrophy. Proceedings of the National Academy of Sciences of the United States of America, 2009, 106 (33), pp.13915-13920. ⟨10.1073/pnas.0905780106⟩. ⟨hal-03173848⟩
- Arnaud Lafaurie, Nathalie Azéma, Laurent Ferry, José-Marie Lopez-Cuesta. Stability parameters for mineral suspensions: Improving the dispersion of fillers in thermoplastics. Powder Technology, 2009, 192 (1), pp.92-98. ⟨10.1016/j.powtec.2008.11.018⟩. ⟨hal-03181081⟩
- Julien Rouyer, Arnaud Poulesquen, Lionel Desgranges, Cécile Ferry. Modelling of the spent fuel oxidation: toward the operational model. Journal of Nuclear Materials, 2009, 395 (1-3), pp.89-98. ⟨10.1016/j.jnucmat.2009.09.023⟩. ⟨hal-00443739⟩
- Véronique Ducros, Maud Andriollo-Sanchez, Josiane Arnaud, Nathalie Meunier, François Laporte, et al.. Zinc supplementation does not alter plasma homocysteine, vitamin B12 and red blood cell folate concentrations in French elderly subjects.. Journal of Trace Elements in Medicine and Biology, 2009, 23 (1), pp.15-20. ⟨10.1016/j.jtemb.2008.08.003⟩. ⟨inserm-00392277⟩
- Morgane Stum, Emmanuelle Girard, Marie Bangratz, Véronique Bernard, Marc Herbin, et al.. Evidence of a dosage effect and a physiological endplate acetylcholinesterase deficiency in the first mouse models mimicking Schwartz-Jampel syndrome neuromyotonia.. Human Molecular Genetics, 2008, 17 (20), pp.3166-79. ⟨10.1093/hmg/ddn213⟩. ⟨hal-00335149⟩
- Alban Vignaud, Françoise Fougerousse, Etienne Mouisel, Christelle Bertrand, Beatrice Bonafos, et al.. Genetic ablation of acetylcholinesterase alters muscle function in mice.. Chemico-Biological Interactions, 2008, 175 (1-3), pp.129-30. ⟨10.1016/j.cbi.2008.04.035⟩. ⟨hal-00289869⟩
- Arnaud F Klein, Mitsuru Ebihara, Christine Alexander, Marie-Josée Dicaire, A. Marie-Josée Sasseville, et al.. PABPN1 polyalanine tract deletion and long expansions modify its aggregation pattern and expression. Experimental Cell Research, 2008, 314 (8), pp.1652-1666. ⟨10.1016/j.yexcr.2008.02.005⟩. ⟨hal-03821688⟩
- Anne Bigot, Virginie Jacquemin, Florence Debacq-Chainiaux, Gillian Butler-Browne, Olivier Toussaint, et al.. Replicative aging down-regulates the myogenic regulatory factors in human myoblasts. Biology of the Cell, 2008, 100 (3), pp.189-199. ⟨10.1042/BC20070085⟩. ⟨hal-03833741⟩
- Audrey Vignaud, F. Fougerousse, E. Mouisel, N. Guerchet, C. Hourde, et al.. Genetic inactivation of acetylcholinesterase causes functional and structural impairment of mouse soleus muscles. Cell and Tissue Research, 2008, 333, pp.289-296. ⟨10.1007/s00441-008-0640-6⟩. ⟨hal-02659138⟩
- Charlotte Lahoute, Athanassia Sotiropoulos, Marilyne Favier, Isabelle Guillet-Deniau, Claude Charvet, et al.. Premature aging in skeletal muscle lacking serum response factor. PLoS ONE, 2008, 3 (12), pp.e3910. ⟨10.1371/journal.pone.0003910⟩. ⟨hal-02659715⟩
- J. Abraham, P. Abreu, M. Aglietta, C. Aguirre, D. Allard, et al.. Erratum to "Correlation of the highest-energy cosmic rays with the positions of nearby active galactic nuclei" [Astroparticle Physics 29(3) (2008) 188-204]. Astroparticle Physics, 2008, 30, pp.45. ⟨10.1016/j.astropartphys.2008.06.004⟩. ⟨in2p3-00311839⟩
- J. Abraham, P. Abreu, M. Aglietta, C. Aguirre, D. Allard, et al.. Correlation of the highest-energy cosmic rays with the positions of nearby active galactic nuclei. Astroparticle Physics, 2008, 29, pp.188-204. ⟨10.1016/j.astropartphys.2008.01.002⟩. ⟨in2p3-00199474⟩
- Marc Bartoli, J. Poupiot, A. Vulin, F. Fougerousse, L. Arandel, et al.. AAV-mediated delivery of a mutated myostatin propeptide ameliorates calpain 3 but not alpha-sarcoglycan deficiency. Gene Therapy, 2007, 14 (9), pp.733-740. ⟨10.1038/sj.gt.3302928⟩. ⟨hal-01610042⟩
- J. Abraham, M. Aglietta, C. Aguirre, D. Allard, I. Allekotte, et al.. An upper limit to the photon fraction in cosmic rays above 10^19 eV from the Pierre Auger Observatory. Astroparticle Physics, 2007, 27, pp.155-168. ⟨10.1016/j.astropartphys.2006.10.004⟩. ⟨in2p3-00121304⟩
- A. Vignaud, F. Ramond, C. Hourdé, A. Keller, G. Butler-Browne, et al.. Diabetes provides an unfavorable environment for muscle mass and function after muscle injury in mice.. Pathobiology, 2007, 74 (5), pp.291-300. ⟨10.1159/000105812⟩. ⟨hal-00188014⟩
- Françoise Fougerousse, Marc Bartoli, Jérôme Poupiot, Ludovic Arandel, Muriel Durand, et al.. Phenotypic correction of alpha-sarcoglycan deficiency by intra-arterial injection of a muscle-specific serotype 1 rAAV vector. Molecular Therapy, 2007, 15 (1), pp.53-61. ⟨10.1038/sj.mt.6300022⟩. ⟨hal-01610043⟩
- J. Abraham, P. Abreu, M. Aglietta, C. Aguirre, D. Allard, et al.. Correlation of the highest-energy cosmic rays with nearby extragalactic objects. Science, 2007, 318, no 5852, pp.938-943. ⟨10.1126/science.1151124⟩. ⟨in2p3-00188862⟩
- A. Ferry, S. Dugravot, T. Delattre, Christides J.-P., J. Auger, et al.. Identification of a widespread monomolecular odor differentially attractive to several Delia radicum ground dwelling predators in the field.. Journal of Chemical Ecology, 2007, 33, pp.2064-2077. ⟨hal-00193705⟩
- E. Mouisel, B. Blondet, P. Escourrou, Arnaud Chatonnet, J. Molgo, et al.. Outcome of acetylcholinesterase deficiency for neuromuscular functioning.. Neuroscience Research, 2006, pp.389-396. ⟨hal-00109730⟩
- Jose Courty, B. Blondet, G. Carpentier, A. Ferry. Exogenous pleiotrophin applied to lesioned nerve impairs muscle reinnervation.. Neurochemical Research, 2006, 31 (7), pp.907-913. ⟨hal-00109603⟩
- A. Vignaud, Jp Caruelle, I. Martelly, A. Ferry. Differential effects of post-natal development, animal strain and long term recovery on the restoration of neuromuscular function after neuromyotoxic injury in rat.. Comparative Biochemistry and Physiology - Part C: Toxicology and Pharmacology, 2006, pp.1-8. ⟨hal-00109693⟩
- P. Noirez, S. Torres, J. Cebrian, O. Agbulut, J. Pelzer, et al.. TGF-beta1 favors the development of fast type identity during soleus muscle regeneration.. Journal of Muscle Research and Cell Motility, 2006, pp.1-8. ⟨hal-00109688⟩
- E. Mouisel, B. Blondet, P. Escourrou, Arnaud Chatonnet, Jordi Molgó, et al.. Outcome of acetylcholinesterase deficiency for neuromuscular functioning. Neuroscience Research, 2006, 55(4) (4), pp.389-396. ⟨10.1016/j.neures.2006.05.002⟩. ⟨hal-00087591⟩
- V. Mouly, A. Aamiri, Anne Bigot, R. Cooper, S. Di Donna, et al.. The mitotic clock in skeletal muscle regeneration, disease and cell mediated gene therapy. Acta Physiologica Scandinavica, 2005, 184 (1), pp.3-15. ⟨10.1111/j.1365-201X.2005.01417.x⟩. ⟨hal-03833720⟩
- Louis-Philippe Corbeil-Girard, Arnaud F Klein, A. Marie-Josée Sasseville, Hugo Lavoie, Anik Saint-Denis, et al.. PABPN1 overexpression leads to upregulation of genes encoding nuclear proteins that are sequestered in oculopharyngeal muscular dystrophy nuclear inclusions. Neurobiology of Disease, 2005, 18 (3), pp.551-567. ⟨10.1016/j.nbd.2004.10.019⟩. ⟨hal-03832516⟩
- V Jacquemin, D Furling, Anne Bigot, G.S Butler-Browne, V Mouly. IGF-1 induces human myotube hypertrophy by increasing cell recruitment. Experimental Cell Research, 2004, 299 (1), pp.148-158. ⟨10.1016/j.yexcr.2004.05.023⟩. ⟨hal-03833701⟩
- Ana Buj-Bello, Furling D, Helene Tronchere, Jocelyn Laporte, Thierry Lerouge, et al.. Muscle-specific alternative splicing of myotubularin-related 1 gene is impaired in DM1 muscle cells. Human Molecular Genetics, 2002, 11 (19), pp.2297-2307. ⟨10.1093/hmg/11.19.2297⟩. ⟨hal-04481805⟩
- Leïla Haïder, Jacques Tatibouët, Arnaud Lafaurie, Laurent Ferry. In-line ultrasonic characterization of shear dispersion processes of polydisperse fillers in polymer melts. Journal of Physics: Condensed Matter, 2002, 14 (19), pp.4943-4961. ⟨10.1088/0953-8984/14/19/317⟩. ⟨hal-03272075⟩
- Arnaud Ferry, Noirez Philippe, C. Le Page, I. Ben Salah, Dominique Daegelen, et al.. Effects of anabolic/androgenic steroids on regenerating skeletal muscles in the rat. Acta Physiologica Scandinavica, 2001, 166 (2), pp.105-110. ⟨10.1046/j.1365-201x.1999.00549.x⟩. ⟨hal-04588851⟩
- Daniel Skuk, Denis Furling, Jean-Pierre Bouchard, Marlyne Goulet, Brigitte Roy, et al.. Transplantation of Human Myoblasts in SCID Mice as a Potential Muscular Model for Myotonic Dystrophy. Journal of Neuropathology and Experimental Neurology, 1999, 58 (9), pp.921-931. ⟨10.1097/00005072-199909000-00003⟩. ⟨hal-03824113⟩
- Thierry Fauchard, Luc Léger, Serge Berthoin, Georges Cazorla, Yves Eberhard, et al.. Evaluation des aptitudes physiques d'étudiants(e)s en éducation physique de six universités françaises.. STAPS : Revue internationale des sciences du sport et de l'éducation physique, 1999, STAPS : Revue internationale des sciences du sport et de l'éducation physique, 49 (7), pp.8-18. ⟨hal-03481056⟩
- Philippe Noirez, Arnaud Ferry, Imed Ben Salah, Christine Le Page, Juan Pedro Wahrmann, et al.. Effect of increased physical activity on growth and differentiation of regenerating rat soleus muscle. European Journal of Applied Physiology, 1997, 76 (3), pp.270-276. ⟨10.1007/s004210050247⟩. ⟨hal-03564778⟩
- Frédérique Cohet. La valeur explicative de la théorie du patrimoine en droit positif français. RTDCiv. Revue trimestrielle de droit civil, 1996, 4, pp.819. ⟨hal-02111652⟩
Communications dans un congrès
- L. Benarroch, I. Nelson, T. Stojkovic, B Mohand Oumoussa, H. Madry, et al.. P166 Deciphering the genetic cause of oculopharyngodistal myopathy in a French cohort using Cas9-targeted long-read sequencing. 28th International Annual Congress of the World Muscle Society, Oct 2023, Charleston SC, United States. pp.S141, ⟨10.1016/j.nmd.2023.07.298⟩. ⟨hal-04280249⟩
- V. Decostre, C. Chikhaoui, C. Vigouroux, A. Behin, G. Bassez, et al.. VP429 Impaired skeletal muscle strength in adult patients with laminopathies. 28th International Annual Congress of the World Muscle Society, Oct 2023, Charleston SC, United States. pp.S159-S160, ⟨10.1016/j.nmd.2023.07.370⟩. ⟨hal-04280272⟩
- Mário Gomes-Pereira. Toxic RNA and glial cell pathology: deciphering brain dysfunction in myotonic dystrophy. 9th Molecular and Cell Biology Symposium, Feb 2023, Porto, Portugal. ⟨hal-04007409⟩
- Stéphanie Tomé. Long read sequencing in DM1. Workshop on Long read sequencing of expanded tandem repeats, Dec 2022, London, United Kingdom. ⟨hal-04004440⟩
- Laure de Pontual, Guillaume Diop, Sonia Lameiras, Tina Alaeitabar, Sylvain Baulande, et al.. Identification of CTG.CAG contraction factors in Myotonic Dystrophy type 1. 19ème Journées de la Société Française de Myologie (JSFM), Nov 2022, Toulouse, France. ⟨hal-04004435⟩
- Dm Dinca, So Braz, Louison Lallemant, A Gonzalez-Barriga, B Potier, et al.. Glial cell dysfunction in myotonic dystrophy brain disease. 12th Japanese-French Workshop: New insights in personalized medicine for neuromuscular diseases: From Basic to Applied Myology, Sep 2022, Giverny, France. ⟨hal-04004421⟩
- Mário Gomes-Pereira, Arnaud F Klein. Myotonic dystrophy type 1: from DNA repeat expansion and toxic RNA to the development of new therapeutic approaches. European reference network for rare and low prevalence complex diseases, Jul 2022, Webinar, France. ⟨hal-04007414⟩
- Yu-Chih Tsai, Laure de Pontual, Chéryl Heiner, Tanya Stojkovic, Denis Furling, et al.. Identification of a CCG-enriched expanded allele in DM1 patients using Amplification-free long-read sequencing. FASEB, The dynamic DNA structures in biology conference, Jun 2022, Nova Scotia, Canada. ⟨hal-04004408⟩
- Laure de Pontual, Antoine Mangin, Sonia Lameiras, Bin Yang, Tina Alaeitabar, et al.. Identification of CTG.CAG contraction factors in Myotonic Dystrophy type 1. FASEB, The dynamic DNA structures in biology conference, Jun 2022, Nova Scotia, Canada. ⟨hal-04004392⟩
- Laure de Pontual, Antoine Mangin, Bin Yang, Geneviève Gourdon, Vincent Dion, et al.. Identification of CTG.CAG contraction factors in Myotonic Dystrophy type 1. International Myotonic Dystrophy consortium Meeting 13, Jun 2022, Osaka, Japan. ⟨hal-04004375⟩
- Geneviève Gourdon. Overviews of mouse models for myotonic dystrophy. Seminar, Target Laboratory, Nantes, Jun 2022, Nantes (visioconférence), France. ⟨hal-04007452⟩
- Ludovic Arandel, Arnaud F Klein, Frédérique Rau, Alain Sureau, Aurélien Cordier, et al.. Decoy gene therapy to reverse RNA toxicity in DM1. International Myotonic Dystrophy Consortium Meeting IDMC-13, International Myotonic Dystrophy Consortium, May 2022, Osaka, Japan. ⟨hal-03999213⟩
- Charles Frison-Roche, Steve Cottin, Jeanne Lainé, Ludovic Arandel, Marius Halliez, et al.. MBNL loss of function in the motor unit alters neuromuscular communication. International Myotonic Dystrophy Consortium Meeting IDMC-13, International Myotonic Dystrophy Consortium, May 2022, Osaka, Japan. ⟨hal-04005805⟩
- Stéphanie Tomé. How SMRT sequencing can improve the prognosis and genetic counseling in DM1 patients DNA repair. Seminar, May 2022, Cardiff / Virtual, United Kingdom. ⟨hal-04007445⟩
- Yu-Chih Tsai, Laure de Pontual, Cheryl Heiner, T. Stojkovic, Denis Furling, et al.. Identification of a CCG-enriched expanded allele in DM1 patients using Amplification-free long-read sequencing. PacBio users 2022, May 2022, Paris, France. ⟨hal-04004337⟩
- Mário Gomes-Pereira, Arnaud F Klein. Dystrophie myotonique: de la pathophysiologie aux approaches thérapeutiques. Société Française de Thérapie Cellulaire et Génique, Apr 2022, Webinar, France. ⟨hal-04007420⟩
- Geneviève Gourdon. Overviews of mouse models for myotonic dystrophy. Seminar, Askbio, Dec 2021, Web Conference, United States. ⟨hal-04007474⟩
- Laure de Pontual, Antoine Mangin, Bin Yang, Geneviève Gourdon, Vincent Dion, et al.. Identification de nouveaux facteurs de contractions CTG dans la DM1. 18ème Journées de la Société Française de Myologie (JSFM), Nov 2021, Saint Etienne, France. ⟨hal-04004288⟩
- Charles Frison-Roche, Julien Messéant, Jeanne Lainé, Ludovic Arandel, Mégane Lemaitre, et al.. MBNL loss of function in motoneurons leads to motor unit dysfunction in myotonic dystrophy. Journées de la Société Française de Myologie, Société Française de Myologie, Nov 2021, Saint-Etienne, France. ⟨hal-03999311⟩
- Laure de Pontual, Antoine Mangin, Bin Yang, Geneviève Gourdon, Vincent Dion, et al.. Identification of CTG.CAG contraction factors in Myotonic Dystrophy type 1. Expanded repeat disorders: from mechanisms to therapies, Jun 2021, Cardiff, United Kingdom. ⟨hal-04004162⟩
- Stéphanie Tomé, Antoine Mangin, Laure de Pontual, Yu-Chih Tsai, Laetitia Monteil, et al.. How SMRT sequencing can improve the prognosis and genetic counseling in DM1 patient. Expanded repeat disorders: from mechanisms to therapies, Jun 2021, Cardiff, United Kingdom. ⟨hal-04004237⟩
- Stéphanie Tomé. How SMRT sequencing can improve the prognosis and genetic counseling in DM1 patients. PacBio Users, Jun 2021, Web Conference, United States. ⟨hal-04007462⟩
- Mário Gomes-Pereira. Astrocytes, the unusual suspects in myotonic dystrophy. Seminar, Institute for Stem Cell and Exploration of Monogenic Diseases (I-STEM), May 2021, Evry, France. ⟨hal-04007468⟩
- Mário Gomes-Pereira. Myotonic dystrophy: Toxic RNA and glia cell disease. Seminar, Radboud University, May 2021, Radboud, Netherlands. ⟨hal-04007513⟩
- Laure de Pontual, Geneviève Gourdon, Stephanie Tome. Identification of CTG.CAG contraction factors in Myotonic Dystrophy type 1. Société Française de Myologie (SFM), Jan 2021, Paris, France. ⟨hal-04003579⟩
- Geneviève Gourdon. Overviews of mouse models for myotonic dystrophy. Seminar, Biogen, 2021, Web Conference, United States. ⟨hal-04007479⟩
- Geneviève Gourdon. Overviews of mouse models for myotonic dystrophy. Seminar, Bayer AG, Jan 2021, Web Conference, Germany. ⟨hal-04007484⟩
- Stéphanie Tomé, Antoine Mangin, Laure de Pontual, Yu-Chih Tsai, Laetitia Monteil, et al.. How SMRT sequencing can improve the prognosis and genetic counseling in DM1 patients. Neuroscience PacBio day 2020, Dec 2020, Paris, France. ⟨hal-04004354⟩
- Geneviève Gourdon, Stéphanie Tomé. How SMRT sequencing can improve the prognosis and genetic counseling in DM1 patients. Seminar, PacBio Neuroscience day, Dec 2020, Web Conference, United States. ⟨hal-04007488⟩
- Charles Frison-Roche, Julien Messéant, Ludovic Arandel, Mégane Lemaitre, Jeanne Lainé, et al.. MBNL loss of function in motoneurons and motor unit dysfunction in myotonic dystrophy. RNA virtual.. but so real !, Sorbonne Université, Nov 2020, Vidéo, France. ⟨hal-03999245⟩
- A Gonzalez-Barriga, D Dinca, So Braz, Geneviève Gourdon, Mário Gomes-Pereira. Real-time monitoring of brain cell models for neuromuscular and neurodegenerative disorders. Incucyte User Meeting, Nov 2019, Paris, France. ⟨hal-04003539⟩
- Geneviève Gourdon. Overview of DMSXL: pros & cons. Myotonic Annual Conference, Oct 2019, Philadelphia, United States. ⟨hal-04003513⟩
- Geneviève Gourdon. DMSXL mice adults and neonatal features. ENMC International Workshop on Myotonic Dystrophies, Oct 2019, Amsterdam, Netherlands. ⟨hal-04003520⟩
- Mário Gomes-Pereira. Mouse models of brain disease in myotonic dystrophy. ENMC International Workshop on Myotonic Dystrophies, Oct 2019, Amsterdam, Netherlands. ⟨hal-04003506⟩
- Geneviève Gourdon. Overview of the DMSXL mouse model of DM1: From DNA repair to phenotype. Seminar, Institute of Biochemistry and Cell Biology, National Research Council, Sep 2019, Rome, Italy. ⟨hal-04007495⟩
- Moulay Gilles, Lainé Jeanne, Lemaître Mégane, Bitoun Marc, Furling Denis, et al.. Revealing alternative splicing control of clathrin structural plasticity in health and disease. The physics and chemistry of endocytosis at multiple scales, EMBO, Sep 2019, Ischia, Italy. ⟨hal-04007054⟩
- Arnaud F. Klein, Miguel A. Varela, Ludovic Arandel, Ashling Holland, Naira Naouar, et al.. Systemic Delivery of Peptide-Conjugated Antisense Oligonucleotides Leads to Long-Lasting Correction of Myotonic Dystrophy Type I. International Myotonic Dystrophy Consortium Meeting IDMC-12, International Myotonic Dystrophy Consortium, Jun 2019, Gothenburg, Sweden. ⟨hal-03998386⟩
- Denis Furling. RNA-based approaches to reverse repeat expansion toxicity in Myotonic Dystrophy. 6th International Congress of Myology, AFM-Telethon, Mar 2019, Bordeaux, France. ⟨hal-04020148⟩
- So Braz, Aline Huguet-Lachon, C Chhuon, A Schmitt, C Guerrera, et al.. Investigating oligodendrocyte dysfunction in DM1 brain disease. 4th Congress of the Young Researchers of Imagine Institute, May 2018, Paris, France. ⟨hal-04003498⟩
- So Braz, Aline Huguet-Lachon, C Chhuon, A Schmitt, C Guerrera, et al.. Investigating oligodendrocyte dysfunction in DM1 brain disease. International Conference on Unstable Microsatellites and Human Disease, Apr 2018, Capri (IT), Italy. ⟨hal-04003486⟩
- J. Bohm, R. Schneider, E. Malfatti, V. Schartner, X. Lornage, et al.. Integrated analysis of the large-scale sequencing project ``Myocapture'' to identify novel genes for myopathies. 22nd International Congress of the World Muscle Society, Oct 2017, Saint Malo, France. pp.S195, ⟨10.1016/j.nmd.2017.06.367⟩. ⟨hal-01741738⟩
- Ellen L van Agtmaal, Laurane M Andre, Mariekel Willemse, Remco van Cruchten, Ingeborg DGT van Kessel, et al.. Generating Isogenic Myoblast Cell Models by CRISPR/Cas9-mediated Editing, Regulating and Targeting Genes in the Myotonic Dystrophy Type 1 (DM1) Locus. International Myotonic Dystrophy Consortium Meeting IDMC-11, International Myotonic Dystrophy Consortium, Sep 2017, San Francisco, United States. ⟨hal-04019231⟩
- D Dinca, G Sicot, Aline Huguet-Lachon, So Braz, C Chhuon, et al.. CUG RNA toxicity in astrocytes is associated with adhesion and migration deficits and affects neuritogenesis. International Myotonic Dystrophy consortium Meeting 11, Sep 2017, San Francisco (CA, USA), United States. ⟨hal-04003470⟩
- Ludovic Arandel, Micaela Polay-Espinoza, Magdalena Matloka, Audrey Bazinet, Damily de Dea Diniz, et al.. Immortalized Human Myotonic Dystrophy Muscle Cell Lines to Assess Therapeutic Compounds. International Myotonic Dystrophy Consortium Meeting IDMC-11, International Myotonic Dystrophy Consortium, Sep 2017, San Francisco, United States. ⟨hal-04019200⟩
- Diana Dinca, Géraldine Sicot, Aline Huguet-Lachon, Noémie Gueriba, Geneviève Gourdon, et al.. Abnormal neuroglial interactions in myotonic dystrophy. Young Researchers in Life Sciences, May 2017, Paris, France. ⟨hal-04001772⟩
- Geneviève Gourdon. DMSXL mouse model of myotonic dystrophy: past present and future. Seminar, Centre de recherche en myologie, Mar 2017, Paris, France. ⟨hal-04007503⟩
- Diana Dinca, Sandra Braz, Geneviève Gourdon, Mário Gomes-Pereira. Investigating the mechanisms of brain disease in a mouse model of myotonic dystrophy. MDRU meeting, Mar 2017, Munich, Germany. ⟨hal-04001746⟩
- E Dandelot, Stéphanie Tomé, C Dogan, Y Péréon, P Cintas, et al.. Atypical Myotonic Dystrophy type 1 families and CTG repeat contractions. Imagine Young Researcher Seminars, Mar 2017, Paris, France. ⟨hal-04001726⟩
- Sandra Braz, Diana Dinca, Aline Huguet-Lachon, Geneviève Gourdon, Mário Gomes-Pereira. Investigating oligodendrocyte dysfunction in DM1 brain disease. Imagine Young Researcher Seminars, Feb 2017, Paris, France. ⟨hal-04001598⟩
- Alberto Malerba, Pierre Klein, Houria Bachtarzi, Arnaud Ferry, Michael Graham, et al.. Gene replacement therapy as a novel approach for the treatment of oculopharyngeal muscular dystrophy. 45th European Muscle Conference, 2017, Montpellier, France. ⟨hal-04011108⟩
- Florian Britto, Gwénaëlle Begue, Bernadette Rossano, Aurélie Docquier, Barbara Vernus, et al.. REDD1 deletion prevents dexamethasone-induced skeletal muscle atrophy. 12. Day of PhD Students in Chemical and Biological Sciences for Health, May 2014, Montpellier, France. ⟨hal-01837613⟩
- Arnaud Lejeune, Hakim Boudaoud, Norman Mathieu, Michel Potier-Ferry. Automatic solver for computational plasticity basedon Taylor series and automatic differentiation. XII International Conference on Computational Plasticity. Fundamentals and Applications (COMPLAS XII), Jan 2013, Barcelone, Spain. ⟨hal-00983276⟩
- Fabien Béchet, Hakim Boudaoud, Arnaud Lejeune, Michel Potier-Ferry. Résolution de problèmes de contact avec la Méthode Asymptotique Numérique. CFM 2009 - 19ème Congrès Français de Mécanique, Aug 2009, Marseille, France. ⟨hal-03391403⟩
- Hakim Boudaoud, Arnaud Lejeune, Michel Potier-Ferry, Hamid Zahrouni, Isabelle Charpentier. Différentiation automatique et MAN : Application au contact unilatéral. 9e Colloque national en calcul des structures, CSMA, May 2009, Giens, France. ⟨hal-01413186⟩
- A. Ferry, S. Dugravot, Christides J.P., J. Auger, Bagneres A.G., et al.. How to use a general cue to find a specific resource : potential role of volatile concentration in prey location by ground dwelling predators of the cabbage root fly.. 23rd ISCE meeting, Jul 2007, JENA, Germany. pp.268. ⟨hal-00194802⟩
- Arnaud Chatonnet, Christelle Bertrand, Laurence Lepourry, Nathalie Barougier, Anne Ferry. Effect of fluoxetine on neuromuscular function in acetylcholinesterase (AChe) knockout mice. 9.International Meeting on Cholinesterases, May 2007, Suzhou, China. n.p. ⟨hal-02823585⟩
- Audrey Vignaud, F. Fougerousse, E. Mouisel, Christelle Bertrand, Béatrice Bonafos, et al.. Genetic ablation of acetylcholinesterase alters muscle function in mice. 9. International Meeting on Cholinesterases, May 2007, Suzhou, China. ⟨10.1016/j.cbi.2008.04.035⟩. ⟨hal-02758076⟩
- Christelle Bertrand, Béatrice Bonafos, Maud Tremblay, Arnaud Ferry, Arnaud Chatonnet. Effect of fluoxetine on neuromuscular function in acetylcholinesterase (AChE) knockout mice. 9. International Meeting on Cholinesterases, May 2007, Suzhou, China. ⟨10.1016/j.cbi.2008.04.002⟩. ⟨hal-02758520⟩
- E. Mouisel, Arnaud Chatonnet, P. Escourrou, Anne Ferry. Hindlimb muscle contractile properties in acetylcholinesterase knockout mice. Congrès International de Myologie, Jun 2005, Nantes, France. n.p. ⟨hal-02833151⟩
Poster de conférence
- Amélie Vergnol, Alain Sureau, Eric Batsché, Eric Allemand, Massiré Traoré, et al.. CaVβ1A and CaVβ1E embryonic isoforms in adult skeletal muscle, a Mbnl1 related-expression. Myology 2024, Apr 2024, Paris, France. ⟨hal-04782481⟩
- Louison Lallemant, Sandra Braz, Anchel González-Barriga, Paul Magneron, Aurélien Cordier, et al.. Toxic CUG RNA repeats disrupt developmentally-regulated splicing in oligodendrocytes causing transient hypomyelination in a mouse model of myotonic dystrophy.. The European Meeting on Glial Cells in Health and Disease, Jul 2023, Berlin, France. ⟨hal-04005524⟩
- B Potier, Louison Lallemant, Sandrine Parrot, Aline Huguet-Lachon, Geneviève Gourdon, et al.. DM1 transgenic mice exhibit abnormal neurotransmitter homeostasis and synaptic plasticity in association with RNA mis-splicing in the hippocampus.. NeuroFrance, May 2023, Lyon, France. ⟨hal-04005556⟩
- Paul Magneron, Louison Lallemant, Luis Guillermo Correa Parra, Mário Gomes-Pereira, Geneviève Gourdon. Cytoskeleton abnormalities triggered by toxic CUG RNA repeats in DM1 astrocytes.. NeuroFrance, May 2023, Lyon20, France. ⟨hal-04005541⟩
- Alex Corscadden, Louison Lallemant, Hélène Benyamine, Jean-Christophe Comte, Aline Huguet-Lachon, et al.. Defects in mouse cortical glutamate uptake can be unveiled in vivo by a two-in-one quantitative microdialysis.. NeuroFrance, May 2023, Lyon, France. ⟨hal-04005564⟩
- Paul Magneron, Louison Lallemant, Luis Guillermo Correa Parra, Geneviève Gourdon, Mário Gomes-Pereira. Cytoskeleton abnormalities triggered by toxic CUG RNA repeats in DM1 astrocytes.. French Glial Cell Club, May 2023, Lyon, France. ⟨hal-04005531⟩
- Caroline Le Guiner, T Larcher, A Lafoux, G Toumaniantz, S Webb, et al.. Characterization of the muscular and cardiac diseases of the DMSXL mouse model, a transgenic mouse model for Myotonic Dystrophy type 1. American Society of Gene & Cell Therapy, May 2023, LOS ANGELES, United States. ⟨hal-04096181⟩
- Charles Frison-Roche, Steve Cottin, Jeanne Lainé, Ludovic Arandel, Marius Halliez, et al.. MBNL deficiency in spinal motor neurons compromises neuromuscular junction maintenance and gait coordination: implication for Myotonic Dystrophy. Advances in Skeletal Muscle Biology In Health and Disease Conference, Mar 2023, Gainsville, United States. . ⟨hal-05045404⟩
- Amélie Vergnol, Alain Sureau, A. Traoré, X. Lornage, G. Gourdon, et al.. Role of MuscleBlind-Like proteins in the regulation of expression of CaVβ1 isoforms in adult skeletal muscle. 19èmes Journées de la Société Française de Myologie, Nov 2022, Toulouse (FRANCE), France. ⟨hal-03999589⟩
- Eléonore Sizun, Aline Huguet-Lachon, N. Ramanantsoa, Florence Cayetanot, Laurence Bodineau, et al.. Apnées obstructives et mixtes chez le souriceau DMSXL, modèle de la forme congénitale de la maladie de Steinert. Journées de Recherche Respiratoire J2R, Oct 2022, Reims, France. ⟨hal-04009875⟩
- Amélie Vergnol, Alain Sureau, A. Traoré, X. Lornage, G. Gourdon, et al.. Role of MuscleBlind-Like proteins in the regulation of expression of CaVβ1 isoforms in adult skeletal muscle. World Muscle Society 2022, Oct 2022, Halifax (Canada), France. ⟨hal-03999596⟩
- Charles Frison-Roche, Steve Cottin, Jeanne Lainé, Ludovic Arandel, Marius Halliez, et al.. MBNL loss of function in motoneurons leads to motor unit dysfunction in myotonic dystrophy. 7th International Myology Congress, Sep 2022, Nice, France. ⟨hal-04005838⟩
- Xavière Lornage, Michel Ney, Ludovic Arandel, Charles Frison-Roche, Maria Kondili, et al.. MBNL proteins are required for adult skeletal muscle homeostasis and maintenance. 7th International Congress of Myology, Sep 2022, Nice, France. ⟨hal-04000599⟩
- Louison Lallemant, B Potier, Baptiste Grimaud, Sandra Braz, Diana Mihaela Dincã, et al.. Toxic RNA impairs synaptic function and vesicle transport in myotonic dystrophy type 1.. Mechanisms of Neuronal Connectivity Meeting, Sep 2022, New York (NY), France. ⟨hal-04005617⟩
- Yu-Chih Tsai, Laure de Pontual, Cheryl Heiner, Tanya Stojkovic, Denis Furling, et al.. Identification of a CCG-enriched expanded allele in DM1 patients using Amplification-free long-read sequencing.. Myology 2022, Sep 2022, Nice, France. ⟨hal-04005603⟩
- Amélie Vergnol, Alain Sureau, A. Traoré, X. Lornage, G. Gourdon, et al.. Role of MuscleBlind-Like proteins in the regulation of expression of CaVβ1 isoforms in adult skeletal muscle. Myology 2022, Sep 2022, Nice (FRANCE), France. ⟨hal-03999540⟩
- Melinda Gyenge, Catherine Eng, Dalil Hamroun, Teresinha Evangelista, Cynthia Gagnon, et al.. The DM-scope registry: an innovative framework to promote myotonic dystrophy translational research.. 7th International Myology Congress, Sep 2022, Nice, France. ⟨hal-04007065⟩
- Alain Sureau, Aline Huguet, Ludovic Arandel, Aurélien Cordier, Pauline Megalli, et al.. Development of a new mouse model for Myotonic Dystrophy type 1. 7th International Myology Congress, Sep 2022, Nice, France. ⟨hal-04002540⟩
- Julie Tahraoui-Bories, Antoine Mérien, Jeanne Lainé, Florine Roussange, Anchel González-Barriga, et al.. MBNL dependent-impaired development connectivity within neuromuscular circuits in Myotonic Dystrophy type.. Myology 2022, Sep 2022, Nice, France. ⟨hal-04005593⟩
- Germana Falcone, Mariapaola Izzo, Beatrice Cardinali, Jonathan Battistini, Silvia Mandillo, et al.. Application of CRISPR/Cas9 strategy for gene therapy of Myotonic Dystrophy type 1. Myology, Sep 2022, Nice, France. ⟨hal-04009784⟩
- Florent Porquet, L Weidong, K Jehasse, H Gazon, Maria Kondili, et al.. Switch-off the trouble:DMPK promoter targeting by CRISPRi as an original specific therapy in DM1. 7th international congress of Myology, Sep 2022, Nice, France. ⟨hal-04022715⟩
- Yu-Chih Tsai, Cheryl Heiner, Tanya Stojkovic, Denis Furling, Guillaume Bassez, et al.. Nouvelle méthode de Séquençage à longue lecture dans la DM1: vers une caractérisation génotype-phénotype affinée. Conference E-Rare, Sep 2022, Paris, France. ⟨hal-04007021⟩
- Elisabetta Golini, Tiziana Orsini, Mara Rigamonti, Mariapaola Izzo, Jonathan Battistini, et al.. Behavioural abnormalities in DMSXL mice, a model of Myotonic Dystrophy type 1. Federation of European Neuroscience Societies Forum 2022, Jul 2022, Paris, France. ⟨hal-04009698⟩
- Alex Corscadden, Louison Lallemant, Hélène Benyamine, Jean-Christophe Comte, Aline Huguet-Lachon, et al.. Defects in mouse cortical glutamate uptake can be unveiled in vivo by a two-in-one quantitative microdialysis.. Monitoring Molecules in Neuroscience, Jun 2022, Lyon, France. ⟨hal-04006970⟩
- Brigitte Potier, Louison Lallemant, Sandrine Parrot, Aline Huguet-Lachon, Geneviève Gourdon, et al.. DM1 transgenic mice exhibit abnormal neurotransmitter homeostasis and synaptic plasticity in association with RNA mis-splicing in the hippocampus. 18th International Conference - Monitoring Molecules in Neuroscience, Jun 2022, Lyon, France. , ⟨10.3390/ijms2302059⟩. ⟨hal-04006990⟩
- Diana Mihaela Dincã, Louison Lallemant, Anchel González-Barriga, Noemie Cresto, Sandra Braz, et al.. Myotonic dystrophy RNA toxicity alters morphology, adhesion and migration of mouse and human astrocytes. International Myotonic Dystrophy Consortium Meeting, Jun 2022, Osaka, Japan. ⟨hal-04006873⟩
- Yu-Chih Tsai, Laure de Pontual, Cheryl Heiner, Tanya Stojkovic, Guillaume Bassez, et al.. Identification of a CCG-enriched expanded allele in DM1 patients using Amplification-free long-read sequencing.. International Myotonic Dystrophy Consortium Meeting, Jun 2022, Osaka, Japan. ⟨hal-04007014⟩
- Julie Tahraoui-Bories, Antoine Mérien, Florine Roussange, Anchel Gonzalez-Barriga, Jeanne Lainé, et al.. MN dependent-impaired development connectivity within neuromuscular circuits in Myotonic Dystrophy type 1. International Myotonic Dystrophy Consortium Meeting IDMC-13, May 2022, Osaka, Japan. ⟨hal-04022921⟩
- Florine Roussange, Amélie Weiss, Kalina Radoynovska, Jacqueline Gide, Johana Tournois, et al.. Target-agnostic drug discovery approach using informative high-content imaging for identification of a myogenic modulator in DM1 context. International Myotonic Dystrophy Consortium Meeting IDMC-13, May 2022, Osaka, Japan. ⟨hal-04022935⟩
- Florent Porquet, L. Weidong, K. Jehasse, H. Gazon, M. Kondili, et al.. "Switch-off the trouble: DMPK promoter targeting by CRISPRi as an original specific therapy in DM1". International Myotonic Dystrophy Consortium Meeting IDMC-13, May 2022, Osaka, Japan. ⟨hal-04000580⟩
- Cecile Peccate, Chiara D’ercole, Vaarany Karunanuthy, Cecile Bertholle, Brigitte Izac, et al.. Multimodal Single Cell profiling of Duchenne Muscular Dystrophy. EMBO - Muscle formation, maintenance, regeneration and pathology, Apr 2022, Gouvieux, France. ⟨hal-04019214⟩
- Stéphanie Bauché, Damien Sternberg, Céline Buon, Julien Messéant, Myriam Boëx, et al.. Identification of a new splice site mutation in synaptotagmin-2 responsible for a severe and early presynaptic form of congenital myasthenic syndrome. World muscle society, Sep 2021, Online, France. ⟨hal-03994061⟩
- Florent Porquet, L Weidong, K Jehasse, H Gazon, S Blacher, et al.. Highly efficient and specific DMPK promoter inhibition by CRISPRi in DM1 patient-derived myotubes. MDF Annual Conference, Sep 2021, Virtual conference, United States. ⟨hal-04022777⟩
- Lallemant Louison, Gourdon Genevieve, Mário Gomes-Pereira. Study of axonal transport in myotonic dystrophy Type 1. Journée Boris Ephrussi, May 2021, Paris, France. ⟨hal-04009795⟩
- Florent Porquet, L Weidong, K Jehasse, S Blacher, L Massotte, et al.. DMPK promoter silencing by transcriptome editing as a new therapeutic strategy in myotonic dystrophy type 1. 2020 MDF Annual Conference, Sep 2020, Virtual conference, United States. ⟨hal-04022817⟩
- Diana Mihaela Dincã, Anchel González-Barriga, Sandra Braz, Laure-Elise Pillet, Noémie Cresto, et al.. RNA toxicity in myotonic dystrophy causes pronounced spliceopathy in astrocytes, in association with defective cell adhesion and morphology, erratic migration and impaired polarization. Cold Spring Harbor Laboratory Meeting on Glia in Health and Disease, Jul 2020, Cold Spring Harbor (New York), United States. ⟨hal-04009836⟩
- Stéphanie Tomé, Janet Ziegle, Yu-Chih Tsai, John Harting, Jean-Paul Bonnefont, et al.. How SMRT sequencing can improve the prognosis and genetic counseling in DM1 patients. European Society of Human Genetics Annual Meeting, Jun 2020, Online, France. ⟨hal-04009915⟩
- Stéphanie Bauché, Sureau Alain, Damien Sternberg, Céline Buon, Julien Messeant, et al.. Identification of a new splice site mutation in synaptotagmin-2 responsible for a severe and early presynaptic form of congenital myasthenic syndrome. journées de la société française de myologie, Nov 2019, Marseille (13), France. ⟨hal-03994043⟩
- Charles Frison-Roche, Julien Messéant, Ludovic Arandel, Mégane Lemaitre, Frédérique Rau, et al.. Consequences of impaired MBNL function in motoneurons on the motor unit. Journées de la Société Française de Myologie, Nov 2019, Marseille (13), France. ⟨hal-04002524⟩
- Alexandra Monceau, Clément Delacroix, Mégane Lemaitre, Gaelle Revet, Denis Furling, et al.. Prox1 gene transfer combined with voluntary exercise improves dystrophic muscle fragility in Mdx mice.. 17th days of French Society of Myology, Nov 2019, Marseille (FRANCE), France. ⟨hal-04011604⟩
- Florent Porquet, S Ormenese, F Giroulle, L Massotte, S Freeman, et al.. DMPK promoter silencing by CRISPRi as a new therapeutic strategy in myotonic dystrophy type 1. 2019 MDF annual conference, Sep 2019, Philadelphie, United States. ⟨hal-04022843⟩
- Sandra Braz, Raphael Blain, Cyril F Bourgeois, Aline Huguet-Lachon, Alain Schmitt, et al.. Investigating oligodendrocyte dysfunction in DM1 brain disease. The European Meeting on Glial Cells in Health and Disease, Jul 2019, Porto, Portugal. ⟨hal-04010005⟩
- Diana Mihaela Dincã, Anchel González-Barriga, Sandra Braz, Cyril F Bourgeois, Géraldine Sicot, et al.. Toxic RNA affects astrocyte adhesion, spreading and migration in myotonic dystrophy, and impacts neuritogenesis through abnormal glial-neuronal interactions. The European Meeting on Glial Cells in Health and Disease, Jul 2019, Porto, Portugal. ⟨hal-04009975⟩
- Ludovic Arandel, Magdalena Matloka, Arnaud Klein, Joelle Marie, Frédérique Rau, et al.. A Decoy-Based Gene Therapy to Inhibit RNA Toxicity Associated with Expanded CUG. International Myotonic Dystrophy Consortium Meeting IDMC-12, Jun 2019, Gothenburg, Sweden. ⟨hal-04002563⟩
- Anchel González-Barriga, Diana Mihaela Dincã, Sandra Braz, Aurélien Cordier, Cerina Chhuon, et al.. Combination of Omics Approaches to Study Molecular Abnormalities in Individual Brain Cell Types of a DM1 Mouse Model. International Myotonic Dystrophy Consortium Meeting (IDMC-12), Jun 2019, Gothenburg, Sweden. . ⟨hal-04009963⟩
- Aline Huguet-Lachon, Hélène Benyamine, Noémie Gueriba, Pierre David, Elodie Dandelot, et al.. Refinement of the DMSXL mouse phenotype. International Myotonic Dystrophy Consortium Meeting, Jun 2019, Gothenburg, Sweden. ⟨hal-04010043⟩
- Florent Porquet, S Ormenese, F Giroulle, L Massotte, S Freeman, et al.. DMPK promoter silencing by CRISPRi as a new therapeutic strategy in myotonic dystrophy type 1. International Myotonic Dystrophy Consortium Meeting IDMC-12, Jun 2019, Gothenburg, Sweden. ⟨hal-04022900⟩
- B Potier, Aline Huguet-Lachon, Geneviève Gourdon, Mário Gomes-Pereira, Patrick Dutar. Glutamate dysfunction at extrasynaptic site in hippocampus of mice model of myotonic dystrophy type 1 (DM1) disease. NeuroFrance, May 2019, Marseille, France. ⟨hal-04009926⟩
- Delphine Trochet, Bernard Prudhon, Arnaud Ferry, Marc Bitoun. Allele-specific silencing therapy for Dynamin 2-related dominant centronuclear myopathy. Myology, Mar 2019, Bordeaux, France. 2019. ⟨hal-03968190⟩
- Maximilien Sochala, Raphaël Porcher, Tanya Stojkovic, Henri Marc Bécane, Anthony Béhin, et al.. High Risk of Fatal and Non-Fatal Venous Thromboembolism in Myotonic Dystrophy. 6th International Congress of Myology, Mar 2019, Bordeaux, France. ⟨hal-04020135⟩
- Mirella Lo Scrutado, Karine Poulard, C. Sourd, Stéphanie Tomé, Arnaud F Klein, et al.. In vivo genome editing mediates resection of the CTG repeat expansion and decreases pathological signs in a mouse model of myotonic dystrophy type 1. 6th International Congress of Myology, Mar 2019, Bordeaux, France. ⟨hal-04020115⟩
- Ashling Holland, Arnaud F Klein, Miguel Varela, R. Raz, Mike J Gait, et al.. Peptide-PMO as a Therapeutic Modulator for Myotonic Dystrophy Type 1. 6th International Congress of Myology, Mar 2019, Bordeaux, France. ⟨hal-04000595⟩
- Ashling Holland, Arnaud F Klein, Miguel Varela, R. Raz, Mike Gait, et al.. Global Analysis of Peptide-PMO as a Therapeutic Modulator for Myotonic Dystrophy Type 1. 6th International Congress of Myology, Mar 2019, Bordeaux, France. ⟨hal-04020127⟩
- Magdalena Matloka, Ludovic Arandel, Arnaud Klein, Michel Ney, Alain Sureau, et al.. A decoy-based gene therapy targeting CUGexp-DMPK transcripts in DM1. 6th International Myology Congress, Mar 2019, Bordeaux, France. ⟨hal-04002587⟩
- Arnaud F. Klein, Miguel A. Varela, Ludovic Arandel, Ashling Holland, Naira Naouar, et al.. Low-dose of peptide-conjugate antisense oligonucleotides targeting CUGexp-RNA in murine skeletal muscles normalizes Myotonic Dystrophy 1 phenotype. 6th International Congress of Myology, Mar 2019, Bordeaux, France. ⟨hal-04000552⟩
- Elodie Dandelot, Stéphanie Tomé, Céline Guiraud-Dogan, Yann Péréon, Pascal Cintas, et al.. Atypical Myotonic Dystrophy type 1 families and CTG repeats interruptions. Young Researchers of Imagine Institute, May 2018, Paris, France. ⟨hal-04010074⟩
- Stéphanie Tomé, Elodie Dandelot, Céline Guiraud-Dogan, Alexis Bertrand, David Geneviève, et al.. Unusual association of an unique CAG interruption in 5’ of DM1 CTG repeats with intergenerational contraction and low somatic mosaicism. International Conference on Unstable Microsatellites and Human Disease, Apr 2018, Capri, Italy. ⟨hal-04010176⟩
- Elodie Dandelot, Stéphanie Tomé, Geneviève Gourdon. Analyze the role of interruptions in triplet nucleotide repeats diseases: cellular model strategy and Flash-Small-Pool-PCR. International Conference on Unstable Microsatellites and Human Disease, Apr 2018, Capri, Italy. ⟨hal-04010160⟩
- Mirella Lo Scrudato, Karine Poulard, Arnaud F Klein, Stéphanie Tomé, Geneviève Gourdon, et al.. A novel gene editing-based strategy for type 1 myotonic dystrophy. Annual Congress of the World Muscle Society, Oct 2017, Saint Malo, France. ⟨hal-04010235⟩
- Sestina Falcone, C. Benedetto, M Traoré, P. de la Grange, A. Ferry, et al.. From voltage sensing to gene expression in the control of muscle mass homeostasis. 22nd World Muscle Society Congress, Oct 2017, Saint-Malo (France), France. ⟨hal-04002388⟩
- Karim Wahbi, Caroline Chong-Nguyen, Vincent Algalarrondo, Henri Marc Becane, Pauline Arnaud, et al.. Association Between Mutation Size and Cardiac Involvement in Myotonic Dystrophy Type 1. International Myotonic Dystrophy Consortium Meeting IDMC-11, Sep 2017, San Francisco, United States. ⟨hal-04019185⟩
- Damily de Dea Diniz, Alain Sureau, Ludovic Arandel, Frederique Rau, Joelle Marie, et al.. Consequences of ATP2A1 missplicing on muscle function. International Myotonic Dystrophy Consortium Meeting IDMC-11, Sep 2017, San-Fransisco, United States. ⟨hal-03999490⟩
- Ludovic Arandel, Magdalena Matloka, Arnaud Klein, Arnaud Ferry, Frédérique Rau, et al.. A decoy-based gene therapy targeting CUGexp-DMPK transcripts in DM1. International Myotonic Dystrophy Consortium Meeting IDMC-11, Sep 2017, San Fransisco, United States. ⟨hal-04002607⟩
- Stéphanie Tomé, Elodie Dandelot, Céline Guiraud-Dogan, David Geneviève, Yann Péréon, et al.. A single CAG or multiple CCG interruptions within the 5’ end of CTG repeat expansions are associated with contractions across generations and stabilization of the repeat in blood. International Myotonic Dystrophy Consortium Meeting, Sep 2017, San Francisco, United States. ⟨hal-04010211⟩
- Daniel Owens, Julien Messeant, G Herledan, Arnaud Ferry, Anne Bertrand, et al.. Nuclear envelope protein lamin A/C is a crucial mechanosensory component for skeletal muscle plasticity. International Congress of Neuromuscular Disorders, Sep 2017, Ottawa, Canada. 2017. ⟨hal-03968419⟩
- Diana Mihaela Dincã, Géraldine Sicot, Aline Huguet-Lachon, Noémie Gueriba, Geneviève Gourdon, et al.. Impact of the CTG repeat expansion on neuronal and astroglial physiology in myotonic dystrophy. Colloque des Jeunes Chercheurs, Jun 2017, Paris, France. ⟨hal-04010254⟩
- Gonzalo Cordova, Ludovic Arandel, Negroni Elisa, Fanny Roth, Alison Oliver, et al.. Model of human skeletal muscle in immunodeficient mice for the study of human muscle regeneration and therapies for muscle disorders. Colloque Jeunes Chercheurs 2017 - Journées des familles, Jun 2017, Paris, France. ⟨hal-04012483⟩
- Arnaud Klein, Ludovic Arandel, Andrey Arzumanov, Lucile Revillod, Miguel Varela, et al.. Enhanced systemic delivery of antisense oligonucleotides using cell-penetrating peptide to reverse RNA toxicity in DM1. International Myotonic Dystrophy Consortium Meeting IDMC-11, May 2017, San-Fransisco, United States. ⟨hal-03999475⟩
- Diana Mihaela Dincã, Laurent Servais, Géraldine Sicot, Axelle Leroy, Cynthia Prigogine, et al.. Upregulation of glial GLT1 glutamate transporter corrects Purkinje cell dysfunction and cerebellum-dependent motor incoordination in a mouse model of myotonic dystrophy. SAB Meeting, Apr 2017, Paris, France. ⟨hal-04010282⟩
Ouvrages (y compris édition critique et traduction)
- Christophe Bident, Arnaud Maisetti, Sylvie Patron (Dir.). Dans la solitude de Bernard-Marie Koltès. Editions Hermann, 2014, Cahiers Textuel, 978-2705688356. ⟨halshs-02343477⟩
Chapitres d'ouvrage
- Stéphanie Tomé, Geneviève Gourdon. Fast Assays to Detect Interruptions in CTG.CAG Repeat Expansions. Trinucleotide Repeats, 2056, Springer New York, pp.11-23, 2020, Methods in Molecular Biology, ⟨10.1007/978-1-4939-9784-8_2⟩. ⟨hal-03545514⟩
- Arnaud F. Klein, Ludovic Arandel, Joëlle Marie, Denis Furling. FISH protocol for Myotonic Dystrophy type 1 cells. Trinucleotide Repeats Methods and Protocols, 2056, pp.203 - 215, 2019, ⟨10.1007/978-1-4939-9784-8⟩. ⟨hal-02996909⟩
Pré-publications, Documents de travail
- Laureline Bétemps, Stéphane Descorps-Declère, Olivia Frenoy, Lucie Poggi, Valentine Mosbach, et al.. TALEN-induced contraction of CTG trinucleotide repeats in myotonic dystrophy type 1 cells. 2024. ⟨pasteur-04892479⟩
- Diana Dincã, Anchel González-Barriga, Geraldine Sicot, Louison Lallemant, Laure-Elise Pillet, et al.. Myotonic dystrophy RNA toxicity alters morphology, adhesion and migration of mouse and human astrocytes. 2022. ⟨hal-03863376⟩
- Hong Joo Kim, Payam Mohassel, Sandra Donkervoort, Lin Guo, Kevin O’donovan, et al.. Specific heterozygous frameshift variants in hnRNPA2B1 cause early-onset oculopharyngeal muscular dystrophy. 2022. ⟨hal-03790055⟩
Thèses
- Ludovic Arandel. Développement d'une thérapie génique pour la Dystrophie Myotonique de type 1. Génétique humaine. Sorbonne Université, 2023. Français. ⟨NNT : 2023SORUS229⟩. ⟨tel-04238758⟩
- Charles Frison-Roche. Perte des protéines MBNL dans les motoneurones : impact sur la fonction de l’unité motrice dans la Dystrophie myotonique. Médecine humaine et pathologie. Sorbonne Université, 2022. Français. ⟨NNT : 2022SORUS305⟩. ⟨tel-04265649v2⟩
- Alexandra Monceau. Effet de l'exercice physique, combiné ou non à une thérapie génique, sur la fonction musculaire de modèles murins de dystrophie musculaire de Duchenne. Physiologie [q-bio.TO]. Sorbonne Université, 2021. Français. ⟨NNT : 2021SORUS172⟩. ⟨tel-03993727⟩
- Elodie Dandelot. Familles atypiques et rôle des interruptions sur l'instabilité des triplets CTG impliqués dans la Dystrophie Myotonique de type 1 (DM1). Génétique. Université Paris Cité, 2019. Français. ⟨NNT : 2019UNIP5089⟩. ⟨tel-04008729⟩
- Sandra Oliveira Braz. Mechanisms of brain dysfunction in myotonic dystrophy type 1 : impact of the CTG expansion on oligodendrocytes and myelin. Genetics. Université Paris Cité, 2019. English. ⟨NNT : 2019UNIP5086⟩. ⟨tel-03978221⟩
- Magdalena Matloka. MBNL derivatives for therapeutic application in myotonic dystrophy. Molecular biology. Sorbonne Université, 2019. English. ⟨NNT : 2019SORUS269⟩. ⟨tel-03344359⟩
- Damily de Dea Diniz. The study of the consequences of SERCA1’s misplicing on muscle function in Myotonic Dystrophy type 1. Life Sciences [q-bio]. Sorbonne université, 2018. English. ⟨NNT : ⟩. ⟨tel-03995016⟩
- Diana Mihaela Dincã. Mechanisms of brain dysfunction in myotonic dystrophy type 1 : impact of the CTG expansion on neuronal and astroglial physiology. Life Sciences [q-bio]. Sorbonne paris Cité, 2017. English. ⟨NNT : 2017USPCB054⟩. ⟨tel-04008705⟩
Documents récupérés de l'archive ouverte HAL ![]()






L’équipe bénéficie également de partenariats privés.