

Repeat Expansions & Myotonic Dystrophy (REDs)
La thématique principale de l’équipe est centrée sur la Dystrophie Myotonique, une des maladies neuromusculaires les plus fréquentes chez l’adulte, et plus particulièrement sur la DM de type 1 (DM1) également appelée maladie de Steinert. La DM1 est caractérisée par une faiblesse et une atrophie musculaire progressive, une myotonie, des défauts de conduction cardiaque, une cataracte précoce, des troubles endocriniens et gastro-intestinaux ainsi que des atteintes neurologiques. On distingue cinq formes cliniques de cette maladie multisystémique incluant les formes tardive, adulte, juvénile, infantile et congénitale. Actuellement, il n’y a pas de traitement pour cette maladie génétique mais diverses approches thérapeutiques sont en développement.
La DM1 est une maladie autosomique dominante causée par une expansion anormale de triplets CTGn (n>40) localisée dans la région 3’ non codante du gène DMPK. Le caractère instable de cette expansion de séquences CTG répétées se manifeste dans différents tissus tout au long de la vie du patient mais également entre générations successives, et représente la base moléculaire du phénomène d’anticipation observé dans cette maladie. La DM1 est une maladie à gain-de-fonction d’ARN. En effet, les transcrits DMPK mutés contenant des répétitions CUG pathologiques (ARN-CUGexp) sous retenus dans le noyau des cellules sous forme d’agrégats riboprotéiques (foci) et perturbent les fonctions de certaines protéines de liaison aux ARNs (RBPs). Notamment les foci séquestrent les RBPs de la famille MBNL impliquées dans le processus de maturation des ARNs. Ainsi, la perte fonctionnelle de MBNL conduit à des défauts d’épissage alternatif de certains ARN pré-messagers qui ont été associés à des symptômes comme ceux du CLCN1 à la myotonie, de INSR à la résistance à l’insuline, de BIN1 à la faiblesse musculaire, de DMD à l’altération de l’architecture des fibres musculaires et de SCN5A à des défauts de conduction et du rythme cardiaque. Cependant des mécanismes additionnels sont impliqués dans les processus physiopathologiques complexes de cette maladie qui affecte divers types cellulaires et de nombreux tissus.
L’équipe REDs a été créée en 2019 suite à la fusion des équipes de Geneviève Gourdon et de Denis Furling. Elle inclut également le groupe de Guillaume Bassez, neurologue qui coordonne le registre national de dystrophie myotonique (DM-Scope) et celui d’Arnaud Ferry qui s’intéresse à la physiologie musculaire et à l’exercice. L’objectif de cette nouvelle équipe est de synergiser les efforts afin d’accélérer la recherche translationnelle pour cette maladie neuromusculaire et de faire émerger des alternatives thérapeutiques pour les patients. Ainsi les compétences de l’équipe visent à réaliser une recherche intégrée autour de la DM1 allant des mécanismes fondamentaux de l’instabilité des expansions CTG, à la compréhension des mécanismes physiopathologiques à l’aide de modèles cellulaires et animaux, au développement et à l’évaluation d’approches thérapeutiques innovantes et enfin, à la mise en place d’essais pré-cliniques et cliniques pour cette maladie neuromusculaire.
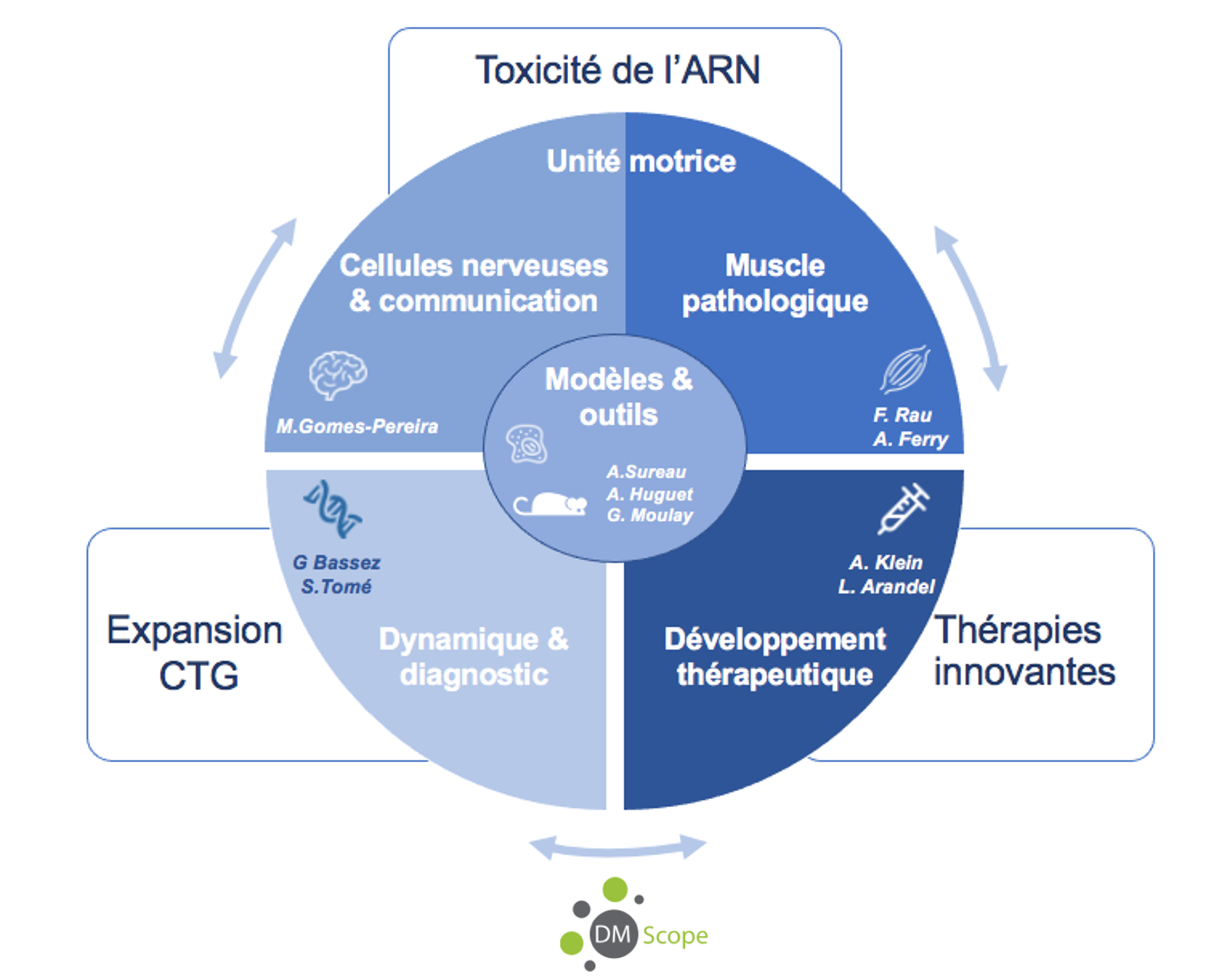
Thèmes de recherche
Dynamique des expansions CTG et implication pour le diagnostic (PIs-G. Bassez & S. Tomé)
A partir de modèle murins et cellulaires nous étudions les mécanismes d’instabilité des répétitions CTG et les facteurs permettant une modulation de leur dynamique. En parallèle, nous étudions des familles DM1 atypiques en lien avec le registre de patients DM-Scope et le service de neuromyologie. Les données croisées entre le laboratoire et la clinique permettrons d’affiner les outils diagnostique et pronostique.
Toxicité de l’ARN et conséquences physiopathologiques
Nous étudions l’impact de la mutation et de l’expression des ARN porteurs d’amplification (ARN-CUGexp) suivant un axe système nerveux/muscle et en particulier sur :
- Les conséquences moléculaires et fonctionnelles sur les différents types de cellules nerveuses et leur communication (PI-M. Gomes Pereira)
- Les motoneurones, l’unité motrice et la fonction musculaire (PIs- F. Rau & A Ferry)
Développement de nouvelles thérapies (PIs : A. Klein & L. Arandel)
Notre équipe développe diverses approches thérapeutiques innovantes pour la DM1, visant différents organes (muscle, cerveau, cœur..) testés à l’aide de modèles cellulaires et murins avec pour objectif le transfert direct vers les services cliniques de l’institut.
Modèles et outils (PIs : A. Surreau , A. Huguet-Lachon & G. Moulay)
De nouveaux modèles cellulaires, murins et outils moléculaires sont développés parallèlement à l’évolution des différents projets.
DM-Scope et recherche clinique (PI: G. Bassez)
- Coordination de l’Observatoire Français des Dystrophies Myotoniques – DM-Scope –
- Caractérisation phénotypique et de l’histoire naturelle de la Dystrophie Myotonique
- Validation d’outils de mesure pour les essais cliniques
- Essais cliniques



Contacts :
| Nom | Position | ORCID | Groupe |
|---|


- Geneviève Gourdon. Overviews of mouse models for myotonic dystrophy. Seminar, Askbio, Dec 2021, Web Conference, United States. ⟨hal-04007474⟩
- Cécile Delorme, Marion Houot, Charlotte Rosso, Stéphanie Carvalho, Thomas Nedelec, et al.. The wide spectrum of COVID-19 neuropsychiatric complications within a multidisciplinary centre. Brain Communications, 2021, 3 (3), ⟨10.1093/braincomms/fcab135⟩. ⟨hal-03464402⟩
- Gilles Moulay, Marc Bitoun, Denis Furling, Steṕhane Vassilopoulos. Comment l’épissage alternatif contribue au contrôle de la plasticité des structures de clathrine. Médecine/Sciences, 2021, 37 (12), pp.1186-1188. ⟨10.1051/medsci/2021178⟩. ⟨hal-03498232⟩
- Laure de Pontual, Antoine Mangin, Bin Yang, Geneviève Gourdon, Vincent Dion, et al.. Identification de nouveaux facteurs de contractions CTG dans la DM1. 18ème Journées de la Société Française de Myologie (JSFM), Nov 2021, Saint Etienne, France. ⟨hal-04004288⟩
- Shira Yanovsky-Dagan, Eliora Cohen, Pauline Megalli, Gheona Altarescu, Oshrat Schonberger, et al.. DMPK hypermethylation in sperm cells of myotonic dystrophy type 1 patients. European Journal of Human Genetics, In press, ⟨10.1038/s41431-021-00999-3⟩. ⟨hal-03449010⟩
- Charles Frison-Roche, Julien Messéant, Jeanne Lainé, Ludovic Arandel, Mégane Lemaitre, et al.. MBNL loss of function in motoneurons leads to motor unit dysfunction in myotonic dystrophy. Journées de la Société Française de Myologie, Société Française de Myologie, Nov 2021, Saint-Etienne, France. ⟨hal-03999311⟩
- Laure de Pontual, Geneviève Gourdon, Stéphanie Tomé. Identification of new factors inducing CTG.CAG repeat contractions in Myotonic Dystrophy type 1. Médecine/Sciences, 2021, 37, pp.6 - 10. ⟨10.1051/medsci/2021182⟩. ⟨hal-03753522⟩
- Alexandra Monceau. Effet de l'exercice physique, combiné ou non à une thérapie génique, sur la fonction musculaire de modèles murins de dystrophie musculaire de Duchenne. Physiologie [q-bio.TO]. Sorbonne Université, 2021. Français. ⟨NNT : 2021SORUS172⟩. ⟨tel-03993727⟩
- Stéphanie Bauché, Damien Sternberg, Céline Buon, Julien Messéant, Myriam Boëx, et al.. Identification of a new splice site mutation in synaptotagmin-2 responsible for a severe and early presynaptic form of congenital myasthenic syndrome. World muscle society, Sep 2021, Online, France. ⟨hal-03994061⟩
- Florent Porquet, L Weidong, K Jehasse, H Gazon, S Blacher, et al.. Highly efficient and specific DMPK promoter inhibition by CRISPRi in DM1 patient-derived myotubes. MDF Annual Conference, Sep 2021, Virtual conference, United States. ⟨hal-04022777⟩
Documents récupérés de l'archive ouverte HAL ![]()






L’équipe bénéficie également de partenariats privés.